
Abstract
Purpose of Review
Angiotensin-converting enzyme 2 (ACE2), a specific high-affinity angiotensin II-hydrolytic enzyme, is the vector that facilitates cellular entry of SARS-CoV-1 and the novel SARS-CoV-2 coronavirus. SARS-CoV-2, which crossed species barriers to infect humans, is highly contagious and associated with high lethality due to multi-organ failure, mostly in older patients with other co-morbidities.
Recent Findings
Accumulating clinical evidence demonstrates that the intensity of the infection and its complications are more prominent in men. It has been postulated that potential functional modulation of ACE2 by estrogen may explain the sex difference in morbidity and mortality.
Summary
We review here the evidence regarding the role of estrogenic hormones in ACE2 expression and regulation, with the intent of bringing to the forefront potential mechanisms that may explain sex differences in SARS-CoV-2 infection and COVID-19 outcomes, assist in management of COVID-19, and uncover new therapeutic strategies.
Similar content being viewed by others

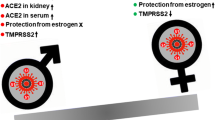

Introduction
Since the first case of coronavirus disease 2019 (COVID-19) was reported in Wuhan, China in December 2019, what was initially a regional epidemic rapidly has expanded into a global pandemic affecting more than 180 countries and territories, bringing significant morbidity and mortality [1]. Though everyone is vulnerable to this disease, certain populations, including those greater than 65 years of age and persons with comorbidities, are more susceptible to infection, present with more severe symptoms, and have worse clinical outcomes. Importantly, available clinical data show that approximately 15% to 30% of the COVID-19 patients are with hypertension and 2.5% to 15% are with coronary heart disease [2,3,4]. Although hypertensive and coronary heart disease patients are often effectively medically managed for their underlying cardiovascular disease conditions, these patients remain prone to develop cardiovascular complications from SARs-Cov2 infection, including arrhythmias, myocarditis, unstable coronary syndrome, and venous and arterial thromboses [5,6,7]. Hypertensive and coronary heart disease patients are also demonstrating higher mortality from COVID-19 than patients without these pre-existing diseases [8].
Male gender is emerging as an additional risk factor for severe COVID-19 and worse outcomes, independent of age. Data from China documents that 54.3–57.3% of hospitalized patients and 61.1% of ICU patients were male [3, 9]. Italian data also show that a significantly greater number of males were admitted to inpatient care compared to females (82% vs 18%) [10]. Zhou et al. [11] reported that 62% of in-hospital deaths in Wuhan were male; similar data from Korea [12] reports 61.1% of in-hospital deaths were male. Considering that the male inpatient number was higher than females in Wuhan, the mortality rate was calculated as 32% (38/119) in males compared to 22% (16/72) in females [11]. In another study of 44,672 individuals with confirmed COVID-19 in Wuhan, the death rate among men was 2.8% compared to 1.7% among women [13, 14]. Likewise, Italy’s case fatality rate as of mid-March 2020, according to the country’s national health institute, was 10.6% in men compared to 6% in women [15]. Identification of sex-specific differences in COVID-19 pathogenesis and disease presentation and intensity could help in directing limited hospital and health care resources and treatments and inform drug development and design.
The reasons for the higher male sex-specific COVID-19-related mortality are likely to be multi-fold, including differences in lifestyle (e.g., higher rates of tobacco smoking and alcohol consumption) and innate immunity [16]. Both Asian and European data document that comorbidities increase the risk for hospitalization and death in COVID-19 patients, and in one study, COVID-19-positive males had more comorbidities than females (59.8% males vs. 40.2% females) [9]. Due to limited patient numbers, however, the prevalence of specific comorbidities varies in different reports [3, 9,10,11,12]. A meta-analysis of 46,248 participants showed that the most prevalent comorbidities among COVID-19-positive hospitalized patients were hypertension (17 ± 7, 95% CI 14–22%), diabetes (8 ± 6, 95% CI 6–11%), cardiovascular disease (CVD; 5 ± 4, 95% CI 4–7%), and respiratory system disease (2 ± 0, 95% CI 1–3%) [17]. Potential differences in these comorbidities as a function of sex have not been explored consistently.
Based on the growing evidence suggesting a sex-based difference in COVID-19 outcomes, an assessment of sex-specific hormone activity, particularly estrogen, in COVID-19 pathogenesis is warranted. Estrogen is known to modulate CVD risk and we [11] and others [18] have established a role for estrogen in regulating renin-angiotensin system (RAS) expression and activity [18,19,20,21,22,23,24,25,26,27,28]. The identification of angiotensin-converting enzyme-2 (ACE2) as the host cell receptor for the SARS-CoV-1 and SARS-CoV-2 [29] coronaviruses has brought attention to the functions of this enzyme outside the domain of its now established role in modulating angiotensin II (Ang II) metabolism as part of the RAS [30]. ACE2 binds the spike protein on the viral capsid [31], which stimulates clathrin-dependent endocytosis [32], the key event in SARS-CoV-2 infection. Here, we discuss the potential connections between SARS-CoV-2 infection and COVID-19 outcomes and sex-specific differences in ACE2, cardiovascular comorbidities, and estrogen activity.
The Renin-Angiotensin System (RAS)
Current knowledge of the biochemical physiology of the RAS has identified an internal arm of the system that acts to limit Ang II pleiotropic actions in the regulation of body fluids, arterial pressure, and cell growth (Fig. 1). In this arm of the system, ACE2 hydrolyzes Ang II to form the heptapeptide angiotensin-(1-7) [Ang-(1-7)], which binds to the Mas receptor (Mas-R) to convey its biological actions. ACE2, functioning as a mono-carboxypeptidase, degrades angiotensin I (Ang I) into angiotensin-(1-9) [Ang-(1-9)], which also possesses antihypertensive properties [33]. The opposite arm of the RAS comprises the ACE enzyme, the effector peptide Ang II, and the receptors AT1-R and AT2-R [34, 35]. Activation of AT1-R by Ang II leads to vasoconstriction and local inflammatory, oxidative stress, proliferative, and profibrotic processes in a large host of organs/tissues including the heart, kidney, lungs, brain, and adipose tissues [36, 37]. The relevance of AT1 activation underlies the development of Ang II receptor blockers (ARBs) as pharmaceutical therapeutics for hypertension, heart failure (HF), diabetic nephropathy, and other CVDs. The vasoactive peptidase angiotensin-converting enzyme (ACE), also a target for therapeutic intervention, catabolizes the decapeptide Ang I into Ang II and, albeit with lower affinity, Ang-(1-9) into Ang-(1-7). ACE inhibitors have been mainstays in the treatment of hypertension and CVD progression for decades [38]. Thus, the two arms of the RAS–ACE/Ang II/AT1-R and ACE2/Ang-(1–7)/Mas-R–have opposing cardiovascular effects. The intrinsic counter-regulatory mechanisms of local tissue RAS are further modulated by a diverse set of autocrine and paracrine factors, including hormones such as estrogen. Recently, a noncanonical pathway leading to Ang II formation, via the conversion of Ang I or angiotensin-(1-12) [Ang-(1-12)] by chymase, was deemed to be the primary contributor in humans to tissue Ang II-induced CVD sequela [39,40,41].

Overview of angiotensinogen products and their functions in the RAS. Angiotensinogen is hydrolyzed into biologically active products [Angiotensin II and Angiotensin-(1-7)] by renin/ACE/CHY/NEP enzymatic pathways. Angiotensin II and Angiotensin-(1-7) interact with receptors present on the membrane (AT1-R and Mas-R, respectively) for their biological activities. Research shows that the spike protein on the capsid of SARS-CoV-2 also binds with ACE2. Abbreviations: CHY, chymase; ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme-2; NEP, neprilysin (neutral endopeptidase 24.11); AT1-R, Angiotensin II type 1 receptor; and Mas-R, Mas receptor
Emphasis on the major contribution that ACE2 has in Ang II metabolism has obscured the fact that the enzyme has hydrolytic activity on des-Ar9-bradykinin, the natural ligand for the bradykinin B1 receptor, as well as apelin-13 and dynorphin A [42,43,44]. Transgenic mice with increased ACE2 expression demonstrate severe, progressive conduction and rhythm disturbances with sustained ventricular tachycardia and terminal ventricular fibrillation. The arrhythmic events are related to the downregulation of the gap junction proteins connexin40 and connexin43 [45]. These data are important because emerging evidence demonstrates the frequent occurrence of cardiac events including arrhythmias and sudden death in patients with advanced COVID-19 infection [46]. The potential interplay between ACE2 and connexins remains to be investigated.
ACE2 in Cardiovascular Disease
Given that ACE2 is widely expressed on the surface of vascular endothelial cells and epithelial cells of the lung, heart, kidney, testis, and the gastrointestinal tract [47,48,49,50], its link to cardiovascular pathology cannot be ignored. In healthy individuals without apparent CVD, circulating ACE2 activity is low, in part due to the presence of a circulating ACE2 inhibitor [51, 52]. Patients with cardiac disease have elevated circulating levels of ACE2 [53]; this is thought to represent either a counter-regulatory mechanism that protects against dysfunction or a factor in the pathogenesis of the disease itself [54], such that release of ACE2 into the circulation and subsequent inhibition decreases its availability in the heart, thereby exacerbating local Ang II effects. Clinical studies show that elevated circulating ACE2 levels predict adverse events in the presence of coronary artery disease [55, 56], atrial fibrillation [57], heart transplantation [58], and HF [59]. In a recent study involving cardiac surgery patients for aortic valve replacement due to aortic stenosis, Ramchand et al. [60] found that elevations in plasma ACE2 activity were associated with reduced myocardial ACE2 gene expression, increased myocardial structural abnormalities such as LV mass and valvular calcification, and more severe cardiac fibrotic remodeling. With a median survival follow-up of 5 years, this study further showed that elevated plasma ACE2 predicted all-cause mortality, irrespective of baseline clinical, imaging, and biochemical findings. While circulating ACE2 levels were higher among male patients, there was no sex-specific difference in overall survival. Moreover, in 161 patients with essential hypertension and 47 age- and sex-matched normotensive healthy subjects, Li et al. [61] reported that serum ACE2 concentrations were positively associated with left atrial diameter, left ventricular end-diastolic diameter, and left ventricular mass in hypertensive patients. Urinary ACE2 levels also positively correlated with systolic blood pressure (SBP) in hypertensive patients [62]. Any effect of inhibition of Ang II synthesis or activity on tissue ACE2 expression/activity was not addressed in these clinical studies. Ferrario’s laboratory first reported in rodents that chronic treatment with ACE inhibitors and ARBs augments ACE2 gene expression and activity in the heart and vasculature of several models of hypertension or cardiac dysfunction [63,64,65]. ACE2 upregulation by ARBs was corroborated by others using mouse and rat models of pressure overload-induced hypertrophic remodeling [66, 67]. These results, when taken together, suggest that elevations in circulating ACE2 are directly linked to clinical abnormalities in cardiovascular function and structure, and in some cases, inversely related to tissue ACE2.
Various preclinical models of CVD also point toward ACE2 as a regulator of end-organ dysfunction, evidenced by relatively lower tissue levels in most cases and higher peripheral ACE2 compared to disease-free counterparts. Kidney ACE2 mRNA and protein expression are substantially lower in spontaneously hypertensive rats (SHR) than in normotensive Wistar-Kyoto (WKY) rats [68]. Pendergrass et al. [69] also found that hypertensive male mRen2.Lewis rats had lower renal cortical ACE2 activity than normotensive Lewis rats. Crackower et al. [70] landmark study revealed that genetic depletion of Ace2 in mice resulted in cardiac systolic dysfunction and an upregulation of cardiac hypoxia-induced genes, along with increases in cardiac, kidney, and circulating Ang II. ACE2 deletion models have a significantly higher mortality rate after myocardial infarction (MI) than wildtype mice, with adverse ventricular remodeling and worsening ventricular function following MI [71]. Trask et al. [72] showed in male (mRen2)27 transgenic hypertensive rats that chronic inhibition of ACE2 with MLN-4760 for 28 days caused accumulation of cardiac Ang II, worsening of cardiac hypertrophy, and fibrosis without changes in circulating Ang II and Ang-(1-7). ACE2 inhibition by MLN-4760 also led to worsening of kidney function in diabetic mice [73].
Sex-based Differences in ACE2
Results from both clinical and laboratory studies underscore a substantial gap in knowledge of sex differences in ACE2 or the impact of sex hormones on the ACE2/Ang-(1-7)/Mas-R axis. In addition to its regulatory role in various CVD states, the existence of a sex-specific pattern of ACE2 expression and activity in cardiovascular tissue may shed light on the observed sexual dimorphic pattern of COVID-19 severity. Sex differences in ACE2 may be due to differences in sex chromosome dosage and/or differences in the gonadal sex hormone milieu. The ACE2 gene is located on the X-chromosome [74]; sex differences in ACE2 expression levels between males and females may thus be dependent on the number of X chromosomes present (one in males and two in females) and the intricacies connected to X-chromosome silencing [75]. Importantly, in an elegant study by Liu and colleagues [75] using the FCG mouse model, higher ACE2 activity in males was found to be a function of the hormonal environment rather than sex chromosome number. Specifically, ACE2 activity in the kidneys of intact mice, gonadectomized mice, and gonadectomized mice given estrogen revealed that only the estrogen-replete group had reduced ACE2 activity, irrespective of the chromosome complement. Interestingly, heart and lung ACE2 activity showed no differences with respect to hormone status or chromosome complement in this experimental model. Oudit et al. [76] found that ACE2 gene knockdown led to increased renal injury susceptibility in male mice, while females were protected. A sexual dimorphic pattern in Ang II-induced kidney disease progression was also reported in ACE2-null diabetic male mice compared to their diabetic female counterparts [77]. Whether sex differences in ACE2 expression, as a function of chromosomal differences, can explain, in part, the sexual dimorphic pattern in Covid-19 epidemiology, irrespective of estrogen status remains speculative.
In addition to the kidney, tissue-specific regulation of ACE and ACE2 in the heart by sex hormones is thought to contribute to sex-related differences in CVD [69, 78, 79]. Using SHRs, Dalpiaz et al. [79] showed higher ACE and ACE2 catalytic activity, lower contractility (dP/dt max), and greater hypertrophy (LV/TL) in males compared to age-matched females. However, following gonadectomy, these sex-related differences were reversed. Orchiectomized males exhibited a decrease in cardiac ACE2 and ACE enzymatic activity and hypertrophy, and a relative increase in contractility compared to intact males. Ovariectomized (OVX) females showed increases in cardiac ACE2 activity without altering ACE activity, and hypertrophic remodeling with reductions in dP/dtmax and concomitant increases in PLB/SERCa2, a marker of reduced cardiomyocyte availability intracellular Ca2+, compared to their gonad-intact counterparts [79]. In the mRen2.Lewis rat, a tissue renin model of hypertension, males exhibited significant increases in cardiac hypertrophy (defined by heart weight normalized to body weight), systolic blood pressure, and cardiac ACE2 activity compared with female littermates [69]. Whether the augmentation in ACE2 activity represents a compensatory effect remains unclear since a sex-related differential in cardiac angiotensins or ACE activity was not observed. Importantly, in normotensive Lewis rats, cardiac ACE2 activity was similar between sexes [69].
Estrogen Regulation of ACE2
Sexual dimorphic patterns in COVID-19 epidemiology and disease severity may be linked to estrogen and estrogen receptor regulation of tissue ACE2. The three naturally occurring estrogens in females are estrone (E1), estradiol (E2), and estriol (E3) [80]. A fourth form of estrogen, estetrol (E4), is produced only during pregnancy [81]. The biological actions of estrogens are mediated by three estrogen receptors (ER), ER[alpha], ER[beta], and G protein-coupled ER (GPER). All three receptors have been localized in cardiovascular, respiratory, nervous, reproductive, and muscle tissue [82,83,84]. The ligand-dependent classical mechanism of estrogen action, mediated by ER[alpha] and ER[beta], involves direct and indirect genomic signaling pathways that result in target gene expression [85, 86]. By contrast, membrane-bound GPER is primarily responsible for the rapid nongenomic actions of estrogens mediated through various protein-kinase cascades [86]. GPER expression is independent to that of the ERs. While estradiol is the primary ligand for the three estrogen receptors, specific agonists including propyl pyrazole triol (PPT), a selective agonist for ER[alpha]; diary propionitrile (DPN), which is a potent ER[β] agonist; and G1,1-[4-(6-bromobenzo[1,3]dioxol-5yl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c] quinolin-8-yl]-ethenone), a GPER agonist that displays no activity at ER[α] and ER[β], are commonly used in preclinical studies to determine the exact roles the individual ERs play in regulating biological functions of various organ systems through health and disease [83, 87].
Using various normotensive and hypertensive rodent models, Groban’s laboratory reported that loss of ovarian estrogen by OVX leads to diastolic dysfunction and LV interstitial remodeling and hypertrophy [22, 88, 89]. Upon probing the local cardiac RAS in normotensive WKY rats and SHRs, we found both strain and estrogen effects with respect to cardiac ACE2 activity [20]. Specifically, ACE2 activity was 25% higher in gonadal-intact normotensive WKY rats versus gonadal-intact SHRs and the loss of ovarian estrogens led to a marked reduction in activity of ACE2 by nearly 30% in the normotensive WKY heart, compared with a 13% decrease in hypertensive SHR hearts. ACE2 deactivation showed a robust correlation to the diastolic dysfunctional phenotype of SHRs and a modest relationship to worsening filling pressures in WKY rats after OVX. In a follow-up study to determine the exact role of estrogen status on ACE2 in isolated cardiomyocytes [21], we again found that ACE2 activity was higher in normotensive WKY rats, although this strain effect was independent of estrogen status. Indeed, loss of ACE2 has been shown to accelerate maladaptive remodeling after MI [71]. Interestingly, studies conducted in pregnant animals further showed that endogenous E2 has a modulatory effect on ACE2 gene expression and activity in the kidney and uterus [90]. In studies using the DOCA (deoxycorticosterone acetate-salt) hypertensive rat model of salt-sensitive hypertension, Shenoy et al. [91] also reported increased levels of ACE2 in the left ventricle after high-dose E2 treatment. While many of these findings suggest that exogenous estrogen up-regulates ACE2, the expression or activity of the enzyme is likely dependent on the specific pathological condition and species. In a hypertensive model due to tissue renin-overexpression, even though no changes in cardiac ACE2 gene or protein expression were observed despite OVX-induced worsening of hypertension and LV diastolic function, chronic estrogen replacement led to reductions in cardiac ACE2 gene and LV tissue expression levels (Fig. 2a) [19]. A similar downregulation in ACE2 gene expression was also shown in the kidneys of estrogen-replete ApoE−/−OVX mice [23]. Discrepancies across these studies might be explained by differences in ACE2 modulation by endogenous estrogens across ages, the underlying conditions of health and disease, and variable dosing and delivery modalities of exogenous E2.

Myocardial ACE2 in mRen2.Lewis rats and spontaneously hypertensive rat (SHR) models. a ACE2 mRNA (left panel), protein level (middle panel), and immunohistochemistry staining intensity (right panel) in left ventricles of sham and ovariectomized (OVX) mRen2.Lewis rats treated with either vehicle (V) or 17β-estradiol (E2, 36 mg/pellet, 60-day release) for 4 weeks. Adapted from Wang et al. [19]. b ACE2 mRNA (left panel), protein (middle panel), and activity (right panel) in the hearts of female sham and OVX SHRs treated with vehicle or G1 (100 μg/kg/day, s.c. for 4 weeks. Image depicts new and adapted data from da Silva, et al. [87]. c Left panel: cardiac ACE2 mRNA expression in sham and ovariectomized mRen2.Lewis rats treated with vehicle or G1 (100 μg/kg/day, s.c. via osmotic mini-pumps) for 2 weeks. Right panel: cardiac ACE2 mRNA in female mRen2.Lewis rats fed with a normal salt (0.5% sodium; control) diet or a high-salt (4% sodium; HS) diet for 10 weeks beginning at 5 weeks of age, and treated with vehicle or G1 (400 μg/kg/day, s.c. via osmotic mini-pumps) for 2 weeks. Image depicts new data from Wang et al. [92] and Jessup et al. [93]. mRNA and protein were determined by real-time PCR and immunoblot, respectively, and corrected by internal control GAPDH. Values are means ± SEM; n = 5–7/group
Cardiac-specific responses to ACE2 modulation by estrogen might also be dependent on the quantity and distribution of estrogen receptors in cardiac cells and tissues. We and others have shown that ER[β] mRNA expression is nondetectable or very low [82, 87, 94, 95], while GPER is similar [87], higher [96], or lower [82] than ER[α] in tissue and cardiomyocytes of rats, mice, and humans of both sexes, depending upon physiologic and pathologic conditions [97, 98] and methodologies used [99]. In OVX SHRs, chronic activation of GPER by its specific agonist G1 (100 μg/kg/d for 4 weeks) downregulates ACE2 protein expression compared to OVX vehicle-treated rats, while cardiac ACE2 is not affected by equipotent treatment with selective agonists to ER[α] (94 μg/kg/day) and ER[β] (58 μg/kg/day) (Fig. 2b) [87]. Similarly, high-salt fed and OVX mRen2.Lewis female rats chronically treated with G1 (400 μg/kg/day and 100 μg/kg/day, respectively, for 14 days) exhibited reduced cardiac ACE2 mRNA compared to their vehicle-treated counterparts (Fig. 2c) [92, 93]. Interestingly, E2 treatment of cultured atrial tissue rendered from older male patients undergoing cardiac surgery led to a protective shift in the ACE/ACE2 ratio at the mRNA and protein levels that were ER[α]-dependent [100]. In contrast, 3 months of E2 treatment in female APOe-OVX mice, a model of atherosclerosis, led to a downregulation of ACE2 mRNA in kidney that was deemed to be mediated via ER[α], since the effect was lost in counterparts lacking ER[α] [23]. Taken together, how estrogen status influences local ACE2 expression and activity in cardiovascular tissue is likely modulated by a complex array of factors, including the types of estrogen receptors present. Whether an E2/GPER regulatory action on ACE2 in the heart has therapeutic potential in lung and other CV tissues in the context of SARs-CoV2 infection remains to be investigated further.
RAS Blockade and COVID-19 Outcomes
We posit that neurohormonal imbalances in the RAS, and particularly in ACE/ACE2, in the circulation or lung and cardiac tissue due to older age [101], comorbid conditions [57, 61, 102, 103], male sex [104, 105], and estrogen deficiency [100, 101] may modulate severity of COVID-19. The potential impact of chronic RAS blockade on ACE2 levels and thus the virulence of SARS-CoV-2 is thus a critical consideration. Both harmful and beneficial effects of ACE inhibitors and ARBs on COVID-19 outcomes have been recently deliberated [106,107,108,109,110]. Preclinical animal studies show that expression and function of ACE2 are markedly increased following chronic RAS blockade. A landmark study by Ferrario et al. [63] demonstrated increased cardiac ACE2 expression and function in normotensive male rats chronically treated with the ACEi lisinopril or ARB losartan compared to vehicle [63]. Ocaranza et al. [111] showed increased cardiac levels and activity of ACE2 1 week following MI from coronary occlusion in ACE inhibitor-treated rats, while Ishiyama et al. [64] showed increased levels of ACE2 mRNA following coronary occlusion in male rats chronically treated with ARBs. An upregulation of ACE2 gene and/or protein expression was also reported in kidneys from male normotensive rats [112], in hypertensive kidneys from male transgenic Ren-2 rats [113], and in thoracic aortas and carotid arteries from male SHRs [65, 114] medicated with RAS blockers. If RAS blockade similarly upregulates ACE2 in the lung’s bronchial branches, the potential exists for an increase in SARS-CoV-2 entry into alveolar type 2 cells [115], thereby increasing the viral load. In addition, an increase in ACE2 expression in the heart following RAS blockade suggests a potential mechanism underlying the observation of acute myocarditis among patients infected with SARS-CoV-2 [46, 116,117,118,119].
While there are many studies showing that ACE2 is upregulated by chronic RAS blockade, it is worth mentioning that no change in ACE2 with ACEi or ARBs have been reported in only two separate studies [120, 121]. Mixed findings across these studies are most likely due to physicochemical differences of ACE inhibitors and ARBs and to differences in their degree of tissue and cell wall penetration [122].
Whether there are sex-specific differences in the effect of chronic ACE inhibitors or ARB treatment on ACE2 upregulation is not clear. To our knowledge, only four studies have reported the effects of RAS inhibition on tissue ACE2 in female animal models. In female normotensive C57BLKS/J mice, Soler et al. [123] showed an increase in ACE2 expression in kidney arterioles after administration of the ARB telmisartan. In female Sprague Dawley rats subjected to subtotal nephrectomy, ACE inhibition by ramipril ameliorated kidney injury-induced reductions in cortical and medullary ACE2 activity [124]. Myocardial infarction–related increases in cardiac ACE2 gene and protein expression were exacerbated by ACE and ARB inhibition and combination therapy in female Sprague Dawley rats [121]. In contrast, we observed significant reductions in cardiac ACE2 mRNA and activity in OVX SHRs following long-term treatment with the ACE inhibitor lisinopril compared to gonad-intact, OVX-vehicle, and OVX-G1-treated age-matched animals (unpublished data). As expected, ACE inhibition led to marked reductions in blood pressure compared to the other groups [125], which may account for the reductions in ACE2 in this hypertensive model.
In terms of hypertension control, one systematic review of the literature revealed that sex-specific outcome data were only reported in 43% of clinical trials reviewed, with ACE inhibitors and ARBs conveying a slightly higher cardiovascular benefit in men versus women [126]. Reduced blood-pressure-lowering effects of ACE inhibition in females has also been supported in animal studies [127], with ARBs potentially providing more benefit in females. Furthermore, in a retrospective review of nearly 20,000 older (≥ 65 years of age) patients with HF, women on ARBs had better survival than those on ACE inhibitors (adjusted hazard ratio [HR] 0.69, 95% confidence interval [CI] 0.59, 0.80), while no difference in survival was found in men prescribed ARBs compared to ACEi (HR 1.10, 95% CI 0.95, 1.30) [128]. Whether sex differences in efficacy of ACE inhibitors and ARBs in controlling hypertension and HF might contribute to differences in the effect of chronic RAS inhibition on COVID-19 outcomes is not known. With the advent of personalized medicine, the factors considered for a patient with CVD should not only include age, race, concomitant diseases, medications, and renal and hepatic function but also sex. While the RAS plays a key role in the development and progression of CVD, substantial variability in individual responses to ACEi and ARBs exists, particularly with respect to sex. Understanding and identifying genes, gene products, and enzymatic activities involved in the differential responses to RAS inhibitors could improve our knowledge of how drugs differentially affect patients exposed to environmental stressors (e.g., airborne infectious agents such as SARS-CoV-2) by sex [129].
Ironically, RAS inhibition that enhances tissue ACE2 may be a means to protect the heart and lung. ACE2 activity increases the production of Ang-(1-7), which leads to vasodilatory, antiproliferative, antifibrotic, and antithrombotic effects, processes that would counteract the negative cardiovascular effects of SARS-CoV-2 infection. We know that overexpression of ACE2 prevents adverse cardiac remodeling [130], and that treatment with Ang-(1-7) limits cardiac hypertrophy and fibrosis in rodent models subjected to hemodynamic stress [131,132,133]. Pulmonary ACE2 may also have a protective role via regulating the balance of circulating Ang II/Ang-(1-7) levels [134]. It has been postulated that unrestricted angiotensin II actions may, in part, be responsible for organ injury in SARS-CoV-2 infection [135]. Continued viral replication contributes to reduced membrane ACE2 expression in cultured cells [136]. Down-regulation of ACE2 activity in the lungs could facilitate the initial neutrophil infiltration in response to bacterial endotoxin and may result in unopposed angiotensin II accumulation and local RAS activation [42]. While increases in Ang II during hypoxic periods lead to pulmonary vasoconstriction [137], which is important in preventing shunting, it can also trigger increases in vascular permeability that contribute to pulmonary edema [138]. In acute respiratory distress syndrome (ARDS) models, ACE2 knockdown induced more severe symptoms of disease compared to ACE2-intact mice, while overexpression appeared to be protective [138, 139]. The “cytokine storm” described as the ultimate terminal event in ARDS is an example how protective immunological mechanisms can become lethal when the magnitude of the biological response overwhelms control mechanisms [140].
To date, only one study has reported a potential for positive effects of RAS inhibition on COVID-19 outcomes. In a retrospective study of Chinese patients hospitalized with COVID-19 from January 11, 2020 to February 23, 2020, Meng et al. [141] evaluated the medical records of 42 patients (median age 64.5 years; 57.1% male) taking antihypertensive medications. Of these patients, 17 were taking ACE inhibitors or ARBs and the remaining 25 were taking other antihypertensive medications, including calcium channel blockers, beta-blockers, and diuretics. Patients not on ARBs or ACE inhibitors had higher rates of severe infection (48% [n = 12] vs. 23.5% [n = 4]), although the study was too small to detect significance or effectively differentiate outcomes by sex. While no overt benefit from RAS blockade was reported in two large population-based observational studies, one in Lombardy, Italy involving 6272 case patients [142] and one in a large health care network in New York City involving 12,594 tested patients [143], it is important to note that no association between use of ARBs or ACE inhibitors, or any other antihypertensive medications, was confirmed in those found to be positive for Covid-19, nor among those who had a severe or fatal course of the disease. Moreover, no association between drug class and disease presence or severity was found according to sex [8]. Despite the inherent limitations of observational studies, these studies involving separate populations provide some reassurance that the continued use of ACE inhibitors and ARBs is unlikely to be harmful in patients with Covid-19. To realize the exact impact of continuation versus discontinuation of ACE inhibitors and ARBs on outcomes in hospitalized patients with Covid-19, we will need to await findings from the recently initiated randomized clinical trial, REPLACECOVID (NCT04338009). At this time, the authors agree with recommendations from the European Society of Cardiology (https://www.escardio.org/Education/COVID-19-and-Cardiology/ESC-COVID-19-Guidance) and the American College of Cardiology, Heart Failure Society of America and the American Heart Association (https://www.acc.org/latest-in-cardiology/articles/2020/03/17/08/59/hfsa-acc-aha-statement-addresses-concerns-re-using-raas-antagonists-in-covid-19) that patients should be maintained on their usual treatment for blood pressure control and cardiac protection including ACE-inhibitors, ARBs, and any other anti-hypertensive therapies and that treatment should not be withdrawn if Covid-19 symptoms manifest. Indeed, premature withdrawal of these medications may increase the risk of rebound hypertension and/or clinical decompensation in high-risk patients. These societies further remark that initiation of ACE inhibitors and ARBs therapy in very ill and unstable Covid-19 positive patients is not worthwhile.
In summary, the accumulating evidence of a somewhat lower rate of COVID-19 disease severity in women needs to be further investigated and correlated with the therapies that infected women were taking at the time of the event. Large data banks being generated in response to the pandemic need to be probed for sex-driven differences in clinical presentation of the disease, age, and medical history, including records of anticontraceptive use and menopausal hormone therapy. The undeniable evidence regarding the cardio-renal protective role of estrogen and the increased rate of CVD expression post-menopause strongly suggests an influence on the sensitivity of women to SARS-CoV-2 infection. It is worth noting that two clinical trials have been initiated to examine whether short-term treatment of male Covid-19 positive patients with an estrogen patch (NCT04359329) or progesterone (NCT04365127), in an effort to favorably modulate immune system responses and limit symptoms to SARS-Cov-2 infection, is beneficial. We urge an aggressive exploration of the current data to affirm these concepts and serve as a guide for current treatment and the development of new therapies.
References
World Health Organization. Coronavirus world map. https://scitechdaily.com/covid-19-world-map-1610909-confirmed-cases-207-countries-99690-deaths/ (2020).
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–13. https://doi.org/10.1016/s0140-6736(20)30211-7.
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan. China JAMA. 2020;323:1061. https://doi.org/10.1001/jama.2020.1585.
Guan W-j, Ni Z-y, Hu Y, Liang W-h, Ou C-q, He J-x et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 2020;382(18):1708–1720. doi:https://doi.org/10.1056/NEJMoa2002032.
Long B, Brady WJ, Koyfman A, Gottlieb M. Cardiovascular complications in COVID-19. Am J Emerg Med. 2020. https://doi.org/10.1016/j.ajem.2020.04.048.
Mehra MR, Ruschitzka F. COVID-19 illness and heart failure: a missing link? JACC Heart Fail. 2020;8(6):512–4. https://doi.org/10.1016/j.jchf.2020.03.004.
Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020. https://doi.org/10.1093/cvr/cvaa106.
Tadic M, Cuspidi C, Mancia G, Dell'Oro R, Grassi G. COVID-19, hypertension and cardiovascular diseases: should we change the therapy? Pharmacol Res. 2020;158:104906. https://doi.org/10.1016/j.phrs.2020.104906.
Guan WJ, Liang WH, Zhao Y, Liang HR, Chen ZS, Li YM, et al. Comorbidity and its impact on 1590 patients with Covid-19 in China: a nationwide analysis. Eur Respir J. 2020;55:2000547. https://doi.org/10.1183/13993003.00547-2020.
Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy region. Italy JAMA. 2020;323:1574. https://doi.org/10.1001/jama.2020.5394.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62. https://doi.org/10.1016/S0140-6736(20)30566-3.
Korean Society of Infectious D, Korea Centers for Disease C. Prevention. Analysis on 54 mortality cases of coronavirus disease 2019 in the Republic of Korea from January 19 to March 10, 2020. J Korean Med Sci. 2020;35(12):e132. https://doi.org/10.3346/jkms.2020.35.e132.
Johnson M. Wuhan 2019 novel coronavirus - 2019-nCoV. Materials and Methods. 2020;10(10):2867. https://doi.org/10.13070/mm.en.10.2867.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet. 2020;395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5.
Onder G, Rezza G, Brusaferro S. Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA. 2020. https://doi.org/10.1001/jama.2020.4683.
Wenham C, Smith J, Morgan R, Gender, Group C-W. COVID-19: the gendered impacts of the outbreak. Lancet. 2020;395(10227):846–848. doi:https://doi.org/10.1016/S0140-6736(20)30526-2.
Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, et al. Prevalence of comorbidities in the novel Wuhan coronavirus (COVID-19) infection: a systematic review and meta-analysis. Int J Infect Dis. 2020;94:91–5. https://doi.org/10.1016/j.ijid.2020.03.017.
Wang H, Sun X, Ahmad S, Su J, Ferrario CM, Groban L. Estrogen modulates the differential expression of cardiac myocyte chymase isoforms and diastolic function. Mol Cell Biochem. 2019;456(1–2):85–93. https://doi.org/10.1007/s11010-018-03492-6.
Wang H, Jessup JA, Zhao Z, Da Silva J, Lin M, MacNamara LM, et al. Characterization of the cardiac renin angiotensin system in oophorectomized and estrogen-replete mRen2.Lewis rats. PLoS One. 2013;8(10):e76992. https://doi.org/10.1371/journal.pone.0076992.
da Silva JS, Gabriel-Costa D, Wang H, Ahmad S, Sun X, Varagic J, et al. Blunting of cardioprotective actions of estrogen in female rodent heart linked to altered expression of cardiac tissue chymase and ACE2. J Renin-Angiotensin-Aldosterone Syst. 2017;18(3):1470320317722270. https://doi.org/10.1177/1470320317722270.
Ahmad S, Sun X, Lin M, Varagic J, Zapata-Sudo G, Ferrario CM, et al. Blunting of estrogen modulation of cardiac cellular chymase/RAS activity and function in SHR. J Cell Physiol. 2018;233(4):3330–42. https://doi.org/10.1002/jcp.26179.
Groban L, Tran QK, Ferrario CM, Sun X, Cheng CP, Kitzman DW, et al. Female heart health: is GPER the missing link? Front Endocrinol (Lausanne). 2019;10:919. https://doi.org/10.3389/fendo.2019.00919.
Brosnihan KB, Hodgin JB, Smithies O, Maeda N, Gallagher P. Tissue-specific regulation of ACE/ACE2 and AT1/AT2 receptor gene expression by oestrogen in apolipoprotein E/oestrogen receptor-alpha knock-out mice. Exp Physiol. 2008;93(5):658–64. https://doi.org/10.1113/expphysiol.2007.041806.
Brosnihan KB, Li P, Ganten D, Ferrario CM. Estrogen protects transgenic hypertensive rats by shifting the vasoconstrictor-vasodilator balance of RAS. Am J Phys. 1997;273(6):R1908–15. https://doi.org/10.1152/ajpregu.1997.273.6.R1908.
Brosnihan KB, Senanayake PS, Li P, Ferrario CM. Bi-directional actions of estrogen on the renin-angiotensin system. Braz J Med Biol Res. 1999;32(4):373–81. https://doi.org/10.1590/s0100-879x1999000400001.
Brosnihan KB, Weddle D, Anthony MS, Heise C, Li P, Ferrario CM. Effects of chronic hormone replacement on the renin-angiotensin system in cynomolgus monkeys. J Hypertens. 1997;15(7):719–26. https://doi.org/10.1097/00004872-199715070-00003.
Chappell MC, Gallagher PE, Averill DB, Ferrario CM, Brosnihan KB. Estrogen or the AT1 antagonist olmesartan reverses the development of profound hypertension in the congenic mRen2. Lewis rat Hypertension. 2003;42(4):781–6. https://doi.org/10.1161/01.HYP.0000085210.66399.A3.
Gallagher PE, Li P, Lenhart JR, Chappell MC, Brosnihan KB. Estrogen regulation of angiotensin-converting enzyme mRNA. Hypertension. 1999;33(1 Pt 2):323–8. https://doi.org/10.1161/01.hyp.33.1.323.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3. https://doi.org/10.1038/s41586-020-2012-7.
Ferrario CM. ACE2: more of Ang-(1-7) or less Ang II? Curr Opin Nephrol Hypertens. 2011;20(1):1–6. https://doi.org/10.1097/MNH.0b013e3283406f57.
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–90. https://doi.org/10.1007/s00134-020-05985-9.
Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G, et al. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008;18(2):290–301. https://doi.org/10.1038/cr.2008.15.
Ocaranza MP, Jalil JE. Protective role of the ACE2/Ang-(1-9) axis in cardiovascular remodeling. Int J Hypertens. 2012;2012:594361–12. https://doi.org/10.1155/2012/594361.
Ferrario CM, Chappell MC, Tallant EA, Brosnihan KB, Diz DI. Counterregulatory actions of angiotensin-(1-7). Hypertension. 1997;30(3 Pt 2):535–41. https://doi.org/10.1161/01.hyp.30.3.535.
Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1-7) in regulation of cardiovascular function. Am J Physiol Heart Circ Physiol. 2005;289(6):H2281–90. https://doi.org/10.1152/ajpheart.00618.2005.
Bader M, Ganten D. Update on tissue renin-angiotensin systems. J Mol Med (Berl). 2008;86(6):615–21. https://doi.org/10.1007/s00109-008-0336-0.
Rabelo LA, Alenina N, Bader M. ACE2-angiotensin-(1-7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens Res. 2011;34(2):154–60. https://doi.org/10.1038/hr.2010.235.
Gums JG. Use of ACE inhibitors in the treatment of cardiovascular disease. Am Pharm. 1992;NS32(6):62–70. https://doi.org/10.1016/s0160-3450(15)31098-9.
Ahmad S, Varagic J, Groban L, Dell'Italia LJ, Nagata S, Kon ND, et al. Angiotensin-(1-12): a chymase-mediated cellular angiotensin II substrate. Curr Hypertens Rep. 2014;16(5):429. https://doi.org/10.1007/s11906-014-0429-9.
Ahmad S, Varagic J, VonCannon JL, Groban L, Collawn JF, Dell'Italia LJ, et al. Primacy of cardiac chymase over angiotensin converting enzyme as an angiotensin-(1-12) metabolizing enzyme. Biochem Biophys Res Commun. 2016;478(2):559–64. https://doi.org/10.1016/j.bbrc.2016.07.100.
Reyes S, Varagic J, Ahmad S, VonCannon J, Kon ND, Wang H, et al. Novel cardiac intracrine mechanisms based on Ang-(1-12)/chymase axis require a revision of therapeutic approaches in human heart disease. Curr Hypertens Rep. 2017;19(2):16. https://doi.org/10.1007/s11906-017-0708-3.
Sodhi CP, Wohlford-Lenane C, Yamaguchi Y, Prindle T, Fulton WB, Wang S, et al. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am J Physiol Lung Cell Mol Physiol. 2018;314(1):L17–31. https://doi.org/10.1152/ajplung.00498.2016.
Kalea AZ, Batlle D. Apelin and ACE2 in cardiovascular disease. Curr Opin Investig Drugs. 2010;11(3):273–82.
Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277(17):14838–43. https://doi.org/10.1074/jbc.M200581200.
Donoghue M, Wakimoto H, Maguire CT, Acton S, Hales P, Stagliano N, et al. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J Mol Cell Cardiol. 2003;35(9):1043–53. https://doi.org/10.1016/s0022-2828(03)00177-9.
Madjid M, Safavi-Naeini P, Solomon SD, Vardeny O. Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1286.
Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383(Pt 1):45–51. https://doi.org/10.1042/BJ20040634.
Ferrario CM, Varagic J. The ANG-(1-7)/ACE2/mas axis in the regulation of nephron function. Am J Physiol Renal Physiol. 2010;298(6):F1297–305. https://doi.org/10.1152/ajprenal.00110.2010.
Jia HP, Look DC, Shi L, Hickey M, Pewe L, Netland J, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. 2005;79(23):14614–21. https://doi.org/10.1128/JVI.79.23.14614-14621.2005.
Qi F, Qian S, Zhang S, Zhang Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun. 2020;526:135–40. https://doi.org/10.1016/j.bbrc.2020.03.044.
Lew RA, Warner FJ, Hanchapola I, Yarski MA, Manohar J, Burrell LM, et al. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp Physiol. 2008;93(5):685–93. https://doi.org/10.1113/expphysiol.2007.040352.
Rice GI, Jones AL, Grant PJ, Carter AM, Turner AJ, Hooper NM. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension. 2006;48(5):914–20. https://doi.org/10.1161/01.HYP.0000244543.91937.79.
Uri K, Fagyas M, Kertesz A, Borbely A, Jenei C, Bene O, et al. Circulating ACE2 activity correlates with cardiovascular disease development. J Renin Angiotensin Aldosterone Syst. 2016;17(4). https://doi.org/10.1177/1470320316668435.
Patel VB, Clarke N, Wang Z, Fan D, Parajuli N, Basu R, et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. 2014;66:167–76. https://doi.org/10.1016/j.yjmcc.2013.11.017.
Ramchand J, Patel SK, Srivastava PM, Farouque O, Burrell LM. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS One. 2018;13(6):e0198144. https://doi.org/10.1371/journal.pone.0198144.
Ortiz-Perez JT, Riera M, Bosch X, De Caralt TM, Perea RJ, Pascual J, et al. Role of circulating angiotensin converting enzyme 2 in left ventricular remodeling following myocardial infarction: a prospective controlled study. PLoS One. 2013;8(4):e61695. https://doi.org/10.1371/journal.pone.0061695.
Walters TE, Kalman JM, Patel SK, Mearns M, Velkoska E, Burrell LM. Angiotensin converting enzyme 2 activity and human atrial fibrillation: increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Europace. 2017;19(8):1280–7. https://doi.org/10.1093/europace/euw246.
Soler MJ, Batlle M, Riera M, Campos B, Ortiz-Perez JT, Anguiano L, et al. ACE2 and ACE in acute and chronic rejection after human heart transplantation. Int J Cardiol. 2019;275:59–64. https://doi.org/10.1016/j.ijcard.2018.10.002.
Epelman S, Shrestha K, Troughton RW, Francis GS, Sen S, Klein AL, et al. Soluble angiotensin-converting enzyme 2 in human heart failure: relation with myocardial function and clinical outcomes. J Card Fail. 2009;15(7):565–71. https://doi.org/10.1016/j.cardfail.2009.01.014.
Ramchand J, Patel SK, Kearney LG, Matalanis G, Farouque O, Srivastava PM, et al. Plasma ACE2 activity predicts mortality in aortic stenosis and is associated with severe myocardial fibrosis. JACC Cardiovasc Imaging. 2020;13(3):655–64. https://doi.org/10.1016/j.jcmg.2019.09.005.
Li S, Wang Z, Yang X, Hu B, Huang Y, Fan S. Association between circulating angiotensin-converting enzyme 2 and cardiac remodeling in hypertensive patients. Peptides. 2017;90:63–8. https://doi.org/10.1016/j.peptides.2017.02.007.
Furuhashi M, Moniwa N, Mita T, Fuseya T, Ishimura S, Ohno K, et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am J Hypertens. 2015;28(1):15–21. https://doi.org/10.1093/ajh/hpu086.
Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111(20):2605–10. https://doi.org/10.1161/CIRCULATIONAHA.104.510461.
Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43(5):970–6. https://doi.org/10.1161/01.HYP.0000124667.34652.1a.
Igase M, Strawn WB, Gallagher PE, Geary RL, Ferrario CM. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1-7) expression in the aorta of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;289(3):H1013–9. https://doi.org/10.1152/ajpheart.00068.2005.
Kaiqiang J, Minakawa M, Fukui K, Suzuki Y, Fukuda I. Olmesartan improves left ventricular function in pressure-overload hypertrophied rat heart by blocking angiotensin II receptor with synergic effects of upregulation of angiotensin converting enzyme 2. Ther Adv Cardiovasc Dis. 2009;3(2):103–11. https://doi.org/10.1177/1753944708098691.
Wang X, Ye Y, Gong H, Wu J, Yuan J, Wang S, et al. The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang(1-7)-Mas axes in pressure overload-induced cardiac remodeling in male mice. J Mol Cell Cardiol. 2016;97:180–90. https://doi.org/10.1016/j.yjmcc.2016.05.012.
Tikellis C, Cooper ME, Bialkowski K, Johnston CI, Burns WC, Lew RA, et al. Developmental expression of ACE2 in the SHR kidney: a role in hypertension? Kidney Int. 2006;70(1):34–41. https://doi.org/10.1038/sj.ki.5000428.
Pendergrass KD, Pirro NT, Westwood BM, Ferrario CM, Brosnihan KB, Chappell MC. Sex differences in circulating and renal angiotensins of hypertensive mRen(2). Lewis but not normotensive Lewis rats. Am J Physiol Heart Circ Physiol. 2008;295(1):H10–20. https://doi.org/10.1152/ajpheart.01277.2007.
Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–8. https://doi.org/10.1038/nature00786.
Kassiri Z, Zhong J, Guo D, Basu R, Wang X, Liu PP, et al. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail. 2009;2(5):446–55. https://doi.org/10.1161/CIRCHEARTFAILURE.108.840124.
Trask AJ, Averill DB, Ganten D, Chappell MC, Ferrario CM. Primary role of angiotensin-converting enzyme-2 in cardiac production of angiotensin-(1-7) in transgenic Ren-2 hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;292(6):H3019–24. https://doi.org/10.1152/ajpheart.01198.2006.
Soler MJ, Wysocki J, Ye M, Lloveras J, Kanwar Y, Batlle D. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007;72(5):614–23. https://doi.org/10.1038/sj.ki.5002373.
Komatsu T, Suzuki Y, Imai J, Sugano S, Hida M, Tanigami A, et al. Molecular cloning, mRNA expression and chromosomal localization of mouse angiotensin-converting enzyme-related carboxypeptidase (mACE2). DNA Seq. 2002;13(4):217–20. https://doi.org/10.1080/1042517021000021608.
Liu J, Ji H, Zheng W, Wu X, Zhu JJ, Arnold AP, et al. Sex differences in renal angiotensin converting enzyme 2 (ACE2) activity are 17beta-oestradiol-dependent and sex chromosome-independent. Biol Sex Differ. 2010;1(1):6. https://doi.org/10.1186/2042-6410-1-6.
Oudit GY, Herzenberg AM, Kassiri Z, Wong D, Reich H, Khokha R, et al. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am J Pathol. 2006;168(6):1808–20. https://doi.org/10.2353/ajpath.2006.051091.
Clotet-Freixas S, Soler MJ, Palau V, Anguiano L, Gimeno J, Konvalinka A, et al. Sex dimorphism in ANGII-mediated crosstalk between ACE2 and ACE in diabetic nephropathy. Lab Investig. 2018;98(9):1237–49. https://doi.org/10.1038/s41374-018-0084-x.
Komukai K, Mochizuki S, Yoshimura M. Gender and the renin-angiotensin-aldosterone system. Fundam Clin Pharmacol. 2010;24(6):687–98. https://doi.org/10.1111/j.1472-8206.2010.00854.x.
Dalpiaz PL, Lamas AZ, Caliman IF, Ribeiro RF Jr, Abreu GR, Moyses MR, et al. Sex hormones promote opposite effects on ACE and ACE2 activity, hypertrophy and cardiac contractility in spontaneously hypertensive rats. PLoS One. 2015;10(5):e0127515. https://doi.org/10.1371/journal.pone.0127515.
Thomas MP, Potter BV. The structural biology of oestrogen metabolism. J Steroid Biochem Mol Biol. 2013;137:27–49. https://doi.org/10.1016/j.jsbmb.2012.12.014.
Holinka CF, Diczfalusy E, Coelingh Bennink HJ. Estetrol: a unique steroid in human pregnancy. J Steroid Biochem Mol Biol. 2008;110(1–2):138–43. https://doi.org/10.1016/j.jsbmb.2008.03.027.
Hutson DD, Gurrala R, Ogola BO, Zimmerman MA, Mostany R, Satou R, et al. Estrogen receptor profiles across tissues from male and female Rattus norvegicus. Biol Sex Differ. 2019;10(1):4. https://doi.org/10.1186/s13293-019-0219-9.
Tang ZR, Zhang R, Lian ZX, Deng SL, Yu K. Estrogen-receptor expression and function in female reproductive disease. Cells. 2019;8(10). doi:https://doi.org/10.3390/cells8101123.
Luo T, Kim JK. The role of estrogen and estrogen receptors on cardiomyocytes: an overview. Can J Cardiol. 2016;32(8):1017–25. https://doi.org/10.1016/j.cjca.2015.10.021.
Vrtacnik P, Ostanek B, Mencej-Bedrac S, Marc J. The many faces of estrogen signaling. Biochem Med (Zagreb). 2014;24(3):329–42. doi:https://doi.org/10.11613/BM.2014.035.
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–30. https://doi.org/10.1126/science.1106943.
da Silva JS, Sun X, Ahmad S, Wang H, Sudo RT, Varagic J, et al. G-protein-coupled estrogen receptor agonist G1 improves diastolic function and attenuates cardiac renin-angiotensin system activation in estrogen-deficient hypertensive rats. J Cardiovasc Pharmacol. 2019;74(5):443–52. https://doi.org/10.1097/FJC.0000000000000721.
Zhao Z, Wang H, Jessup JA, Lindsey SH, Chappell MC, Groban L. Role of estrogen in diastolic dysfunction. Am J Physiol Heart Circ Physiol. 2014;306(5):H628–40. https://doi.org/10.1152/ajpheart.00859.2013.
Alencar AK, da Silva JS, Lin M, Silva AM, Sun X, Ferrario CM, et al. Effect of age, estrogen status, and late-life GPER activation on cardiac structure and function in the Fischer344xBrown Norway female rat. J Gerontol A Biol Sci Med Sci. 2017;72(2):152–62. https://doi.org/10.1093/gerona/glw045.
Levy A, Yagil Y, Bursztyn M, Barkalifa R, Scharf S, Yagil C. ACE2 expression and activity are enhanced during pregnancy. Am J Physiol Regul Integr Comp Physiol. 2008;295(6):R1953–61. https://doi.org/10.1152/ajpregu.90592.2008.
Shenoy V, Grobe JL, Qi Y, Ferreira AJ, Fraga-Silva RA, Collamat G, et al. 17beta-estradiol modulates local cardiac renin-angiotensin system to prevent cardiac remodeling in the DOCA-salt model of hypertension in rats. Peptides. 2009;30(12):2309–15. https://doi.org/10.1016/j.peptides.2009.09.005.
Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, Groban L. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res. 2012;94(1):96–104. https://doi.org/10.1093/cvr/cvs090.
Jessup JA, Lindsey SH, Wang H, Chappell MC, Groban L. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS One. 2010;5(11):e15433. https://doi.org/10.1371/journal.pone.0015433.
Pugach EK, Blenck CL, Dragavon JM, Langer SJ, Leinwand LA. Estrogen receptor profiling and activity in cardiac myocytes. Mol Cell Endocrinol. 2016;431:62–70. https://doi.org/10.1016/j.mce.2016.05.004.
Tomicek NJ, Miller-Lee JL, Hunter JC, Korzick DH. Estrogen receptor beta does not influence ischemic tolerance in the aged female rat heart. Cardiovasc Ther. 2013;31(1):32–7. https://doi.org/10.1111/j.1755-5922.2011.00288.x.
De Francesco EM, Angelone T, Pasqua T, Pupo M, Cerra MC, Maggiolini M. GPER mediates cardiotropic effects in spontaneously hypertensive rat hearts. PLoS One. 2013;8(8):e69322. https://doi.org/10.1371/journal.pone.0069322.
Mahmoodzadeh S, Eder S, Nordmeyer J, Ehler E, Huber O, Martus P, et al. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. 2006;20(7):926–34. https://doi.org/10.1096/fj.05-5148com.
Nordmeyer J, Eder S, Mahmoodzadeh S, Martus P, Fielitz J, Bass J, et al. Upregulation of myocardial estrogen receptors in human aortic stenosis. Circulation. 2004;110(20):3270–5. https://doi.org/10.1161/01.CIR.0000147610.41984.E8.
Zhang X, Li T, Liu F, Chen Y, Yao J, Li Z, et al. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol Cell. 2019;73(1):130–42 e5. https://doi.org/10.1016/j.molcel.2018.10.020.
Bukowska A, Spiller L, Wolke C, Lendeckel U, Weinert S, Hoffmann J, et al. Protective regulation of the ACE2/ACE gene expression by estrogen in human atrial tissue from elderly men. Exp Biol Med (Maywood). 2017;242(14):1412–23. https://doi.org/10.1177/1535370217718808.
Fernandez-Atucha A, Izagirre A, Fraile-Bermudez AB, Kortajarena M, Larrinaga G, Martinez-Lage P, et al. Sex differences in the aging pattern of renin-angiotensin system serum peptidases. Biol Sex Differ. 2017;8:5. https://doi.org/10.1186/s13293-017-0128-8.
Epelman S, Tang WH, Chen SY, Van Lente F, Francis GS, Sen S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J Am Coll Cardiol. 2008;52(9):750–4. https://doi.org/10.1016/j.jacc.2008.02.088.
Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. 2020;8(4):e21. https://doi.org/10.1016/S2213-2600(20)30116-8.
Hilliard LM, Sampson AK, Brown RD, Denton KM. The “his and hers” of the renin-angiotensin system. Curr Hypertens Rep. 2013;15(1):71–9. https://doi.org/10.1007/s11906-012-0319-y.
Maric-Bilkan C, Manigrasso MB. Sex differences in hypertension: contribution of the renin-angiotensin system. Gend Med. 2012;9(4):287–91. https://doi.org/10.1016/j.genm.2012.06.005.
Bavishi C, Maddox TM, Messerli FH. Coronavirus disease 2019 (COVID-19) infection and renin angiotensin system blockers. JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1282.
Sparks MA, South A, Welling P, Luther JM, Cohen J, Byrd JB, et al. Sound science before quick judgement regarding RAS blockade in COVID-19. Clin J Am Soc Nephrol. 2020;15:714–6. https://doi.org/10.2215/CJN.03530320.
Kow CS, Zaidi STR, Hasan SS. Cardiovascular disease and use of renin-angiotensin system inhibitors in COVID-19. Am J Cardiovasc Drugs. 2020;20:217–21. https://doi.org/10.1007/s40256-020-00406-0.
AlGhatrif M, Cingolani O, Lakatta EG. The dilemma of coronavirus disease 2019, aging, and cardiovascular disease: insights from cardiovascular aging science. JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1329.
Mourad JJ, Levy BI. Interaction between RAAS inhibitors and ACE2 in the context of COVID-19. Nat Rev Cardiol. 2020;17:313. https://doi.org/10.1038/s41569-020-0368-x.
Ocaranza MP, Godoy I, Jalil JE, Varas M, Collantes P, Pinto M, et al. Enalapril attenuates downregulation of angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension. 2006;48(4):572–8. https://doi.org/10.1161/01.HYP.0000237862.94083.45.
Ferrario CM, Jessup J, Gallagher PE, Averill DB, Brosnihan KB, Ann Tallant E, et al. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005;68(5):2189–96. https://doi.org/10.1111/j.1523-1755.2005.00675.x.
Jessup JA, Gallagher PE, Averill DB, Brosnihan KB, Tallant EA, Chappell MC, et al. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am J Physiol Heart Circ Physiol. 2006;291(5):H2166–72. https://doi.org/10.1152/ajpheart.00061.2006.
Igase M, Kohara K, Nagai T, Miki T, Ferrario CM. Increased expression of angiotensin converting enzyme 2 in conjunction with reduction of neointima by angiotensin II type 1 receptor blockade. Hypertens Res. 2008;31(3):553–9. https://doi.org/10.1291/hypres.31.553.
Lukassen S, Chua RL, Trefzer T, Kahn NC, Schneider MA, Muley T et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are predominantly expressed in a transient secretory cell type in subsegmental bronchial branches. Preprint at https://www.biorxiv.org/content/10.1101/2020.03.13.991455v3 (2020).
Inciardi RM, Lupi L, Zaccone G, Italia L, Raffo M, Tomasoni D, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020. https://doi.org/10.1001/jamacardio.2020.1096.
Kim IC, Kim JY, Kim HA, Han S. COVID-19-related myocarditis in a 21-year-old female patient. Eur Heart J. 2020;41:1859. https://doi.org/10.1093/eurheartj/ehaa288.
Dixon DL, Van Tassell BW, Vecchie A, Bonaventura A, Talasaz A, Kakavand H, et al. Cardiovascular considerations in treating patients with coronavirus (COVID-19). J Cardiovasc Pharmacol. 2020;75:359–67. https://doi.org/10.1097/FJC.0000000000000836.
Zeng JH, Liu YX, Yuan J, Wang FX, Wu WB, Li JX, et al. First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection. 2020. https://doi.org/10.1007/s15010-020-01424-5.
Burrell LM, Risvanis J, Kubota E, Dean RG, MacDonald PS, Lu S, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005;26(4):369–75; discussion 22-4. https://doi.org/10.1093/eurheartj/ehi114.
Burchill LJ, Velkoska E, Dean RG, Griggs K, Patel SK, Burrell LM. Combination renin-angiotensin system blockade and angiotensin-converting enzyme 2 in experimental myocardial infarction: implications for future therapeutic directions. Clin Sci (Lond). 2012;123(11):649–58. https://doi.org/10.1042/CS20120162.
Michel MC, Foster C, Brunner HR, Liu L. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacol Rev. 2013;65(2):809–48. https://doi.org/10.1124/pr.112.007278.
Soler MJ, Ye M, Wysocki J, William J, Lloveras J, Batlle D. Localization of ACE2 in the renal vasculature: amplification by angiotensin II type 1 receptor blockade using telmisartan. Am J Physiol Renal Physiol. 2009;296(2):F398–405. https://doi.org/10.1152/ajprenal.90488.2008.
Velkoska E, Dean RG, Burchill L, Levidiotis V, Burrell LM. Reduction in renal ACE2 expression in subtotal nephrectomy in rats is ameliorated with ACE inhibition. Clin Sci (Lond). 2010;118(4):269–79. https://doi.org/10.1042/CS20090318.
Sun X, Wright KN, Hodge HS, Ahmad S, Varagic J, Wang H, et al. Equivalence of G1/GPER monotherapy compared with dual administration of G1 and Lisinopril in preventing diastolic dysfunction due to estrogen loss in SHR. FASEB J. 2019;33:532.5.
Rabi DM, Khan N, Vallee M, Hladunewich MA, Tobe SW, Pilote L. Reporting on sex-based analysis in clinical trials of angiotensin-converting enzyme inhibitor and angiotensin receptor blocker efficacy. Can J Cardiol. 2008;24(6):491–6. https://doi.org/10.1016/s0828-282x(08)70624-x.
Sullivan JC. Sex and the renin-angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol. 2008;294(4):R1220–6. https://doi.org/10.1152/ajpregu.00864.2007.
Hudson M, Rahme E, Behlouli H, Sheppard R, Pilote L. Sex differences in the effectiveness of angiotensin receptor blockers and angiotensin converting enzyme inhibitors in patients with congestive heart failure--a population study. Eur J Heart Fail. 2007;9(6–7):602–9. https://doi.org/10.1016/j.ejheart.2007.02.001.
Jain KK. Personalized management of cardiovascular disorders. Med Princ Pract. 2017;26(5):399–414. https://doi.org/10.1159/000481403.
Grobe JL, Der Sarkissian S, Stewart JM, Meszaros JG, Raizada MK, Katovich MJ. ACE2 overexpression inhibits hypoxia-induced collagen production by cardiac fibroblasts. Clin Sci (Lond). 2007;113(8):357–64. https://doi.org/10.1042/CS20070160.
Chamsi-Pasha MA, Shao Z, Tang WH. Angiotensin-converting enzyme 2 as a therapeutic target for heart failure. Curr Heart Fail Rep. 2014;11(1):58–63. https://doi.org/10.1007/s11897-013-0178-0.
Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM, et al. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1-7). Am J Physiol Heart Circ Physiol. 2007;292(2):H736–42. https://doi.org/10.1152/ajpheart.00937.2006.
Wang LJ, He JG, Ma H, Cai YM, Liao XX, Zeng WT, et al. Chronic administration of angiotensin-(1-7) attenuates pressure-overload left ventricular hypertrophy and fibrosis in rats. Di Yi Jun Yi Da Xue Xue Bao. 2005;25(5):481–7.
Kuba K, Imai Y, Penninger JM. Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol. 2006;6(3):271–6. https://doi.org/10.1016/j.coph.2006.03.001.
Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med. 2020;382(17):1653–9. https://doi.org/10.1056/NEJMsr2005760.
Dijkman R, Jebbink MF, Deijs M, Milewska A, Pyrc K, Buelow E, et al. Replication-dependent downregulation of cellular angiotensin-converting enzyme 2 protein expression by human coronavirus NL63. J Gen Virol. 2012;93(Pt 9):1924–9. https://doi.org/10.1099/vir.0.043919-0.
Kiely DG, Cargill RI, Lipworth BJ. Acute hypoxic pulmonary vasoconstriction in man is attenuated by type I angiotensin II receptor blockade. Cardiovasc Res. 1995;30(6):875–80.
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–6. https://doi.org/10.1038/nature03712.
Kuba K, Imai Y, Rao S, Jiang C, Penninger JM. Lessons from SARS: control of acute lung failure by the SARS receptor ACE2. J Mol Med (Berl). 2006;84(10):814–20. https://doi.org/10.1007/s00109-006-0094-9.
Ye Q, Wang B, Mao J. The pathogenesis and treatment of the “cytokine storm” in COVID-19. J Inf Secur. 2020;80:607–13. https://doi.org/10.1016/j.jinf.2020.03.037.
Meng J, Xiao G, Zhang J, He X, Ou M, Bi J, et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect. 2020;9(1):757–60. https://doi.org/10.1080/22221751.2020.1746200.
Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Renin-angiotensin-aldosterone system blockers and the risk of Covid-19. N Engl J Med. 2020;382:2431–40. https://doi.org/10.1056/NEJMoa2006923.
Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB, et al. Renin-angiotensin-aldosterone system inhibitors and risk of Covid-19. N Engl J Med. 2020;382:2441–8. https://doi.org/10.1056/NEJMoa2008975.
Funding
The research described here was carried out with support from Program Project Grant HL-051952 from the National Heart, Lung, and Blood Institute of the National Institutes of Health (CMF) and grants AG042758 and AG033727 (LG) from the National Institute on Aging, National Institutes of Health.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the writing and editing of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Hypertension and the Heart
Rights and permissions
About this article

Cite this article
Groban, L., Wang, H., Sun, X. et al. Is Sex a Determinant of COVID-19 Infection? Truth or Myth?. Curr Hypertens Rep 22, 62 (2020). https://doi.org/10.1007/s11906-020-01073-x
Published:
DOI: https://doi.org/10.1007/s11906-020-01073-x