
Abstract
Visceral leishmaniasis (VL) is a severe disease with particular endemicity in over 80 countries worldwide. There is no approved human vaccine against VL in the market. This study was aimed at designing and evaluation of a multimeric vaccine candidate against Leishmania infantum through utilization of helper T lymphocyte (HTL) and cytotoxic T lymphocyte (CTL) immunodominant proteins from histone H1, KMP11, LACK and LeIF antigens. Top-ranked mouse MHC-I, MHC-II binders and CTL epitopes were predicted and joined together via spacers. Also, a TLR-4 agonist (RS-09 synthetic protein) and His-tag were added to the N- and C-terminal of the vaccine sequence, respectively. The final chimeric vaccine had a length of 184 amino acids with a molecular weight of 18.99 kDa. Physico-chemical features showed a soluble, highly-antigenic and non-allergenic candidate. Secondary and tertiary structures were predicted, and subsequent analyses confirmed the construct stability that was capable to properly interact with TLR-4/MD2 receptor. Immunoinformatics simulation displayed potent stimulation of T cell immune responses, with particular rise in IFN-γ, upon vaccination with the proposed multi-epitope candidate. In conclusion, immunoinformatics data demonstrated a highly antigenic vaccine candidate in mouse, which could develop considerable levels clearance mechanisms and other components of cellular immune profile, and can be directed for VL prophylactic purposes.
Similar content being viewed by others


Introduction
Leishmaniases are a family of diseases caused by Leishmania protozoan parasites, having different clinical outcomes, from ulcerative cutaneous lesions to life-threatening generalized disease (Desjeux 2004; Meeting WECotCotL, Organization WH 2010). The Leishmania agents are transmitted via female phlebotomine sand flies, with a substantial impact on 350 million individuals in 88 countries worldwide (Akhoundi et al. 2016; Maroli et al. 2013). Visceral leishmaniasis (VL) or kala-azar is a neglected disease with Leishmania donovani and Leishmania infantum as the causative agents, and based on estimated disease burden demonstrates remarkable morbidity and mortality amongst the tropical infectious diseases (Desjeux et al. 2013; Ready 2014). Current VL treatment options are confined to pentavalent antimonials, paramomycin, miltefosine and amphotericin B; however, they are costly and toxic, require long-term administration, have side effects with the progressive risk of drug resistance phenomenon (Freitas-Junior et al. 2012; Solano-Gallego et al. 2017). As well, control strategies on reservoir hosts and vectors are not entirely applicable in endemic areas (Valero and Uriarte 2020). Alternatively, developing vaccines is a safer option for appropriate VL control, comparable to other therapeutic regimens, which elicit durable immunity against the infection (Iborra et al. 2018).
Nowadays, high-throughput vaccine development is facilitated via a close cooperation among immunologists, molecular biologists and chemical engineers (Parvizpour et al. 2020). Our understanding of the host-pathogen interplay has improved with the discovery of newer technologies, opening unprecedented avenues into the field of rational vaccine design (Khatoon et al. 2017). Such outstanding advances include major histocompatibility complex (MHC) molecule structure and restriction, the nature of antigen presentation, T cell receptor (TCR) elucidation as well as identification of cytokines (Djaoud and Parham 2020). An efficacious vaccine candidate would be able to strongly stimulate IFN-γ-releasing Th-1 cells, through antigen presenting cells (APCs), with subsequent activation of macrophages leading to upsurge in reactive oxygen species (ROS) and nitric oxide to combat intracellular amastigotes (Rodrigues et al. 2016). Within this interaction, various antigenic peptides of the pathogen are provided on the surface of APCs by different allelic forms of MHC molecules. Therefore, proper antigen presentation via MHC molecules is a crucial step, which could be exploited to enhance cell-mediated immunotherapies and multi-epitope-based vaccination platforms (Mahida et al. 2015; Matsumura et al. 1992).
Immunoinformatics approach is a novel way to characterize immunogenic B- and T cell epitopes of a particular antigenic molecule for accurate design and engineering of a multi-epitope subunit vaccine model, which could be directed towards activation of host’s humoral and cellular responses (Parvizpour et al. 2020). The vaccine design field for Leishmania parasites focuses on all three forms of the disease due to the conserved molecules in all involved species (Kumari et al. 2008). In the early twentieth century, “leishmanization” was developed as the first applicable procedure in the history of vaccination against leishmaniasis, however, it was subsequently forbidden to be used in human models for safety concerns (Noazin et al. 2008). As well, the immunity conferred by first-generation vaccines such as those in Iran CL and Sudan VL was not significant regarding cutaneous leishmaniasis (CL) and VL, respectively (Rafati et al. 2005; Singh and Sundar 2012). However, next-generation vaccines including DNA and subunit vaccines opened new doors towards improved vaccine design and delivery against leishmaniasis (Singh and Sundar 2012). In this context, several multicomponent vaccines including KSAC, Leish110-f, Leish111-f and the putative Q protein have been shown to provide protective immunity against VL due to L. infantum; however, no such human vaccine have passed the trials towards international markets to date (Chakravarty et al. 2011; Goto et al. 2011; Joshi et al. 2014; Molano et al. 2003).
Previously, several Leishmania antigens were used as vaccine candidates. Histone H1 includes several basic proteins involved in nucleosome stabilization with high immunogenicity being overexpressed in amastigote and promastigote stages of L. major and L. infantum (Carmelo et al. 2002; Galanti et al. 1998; Requena et al. 2000). Kinetoplastid membrane protein 11 (KMP11) is a highly-conserved potent immunogen in Leishmania, expressed in both developmental stages, and the only antigen being detected by the sera of asymptomatic subjects (Fuertes et al. 1999; Jardim et al. 1995). The Leishmania-activated C-kinase (LACK) antigen, expressed in both amastigote and promastigote forms, is the most investigated component in DNA vaccine candidates against both CL and VL (Nagill and Kaur 2011). Also, Leishmania elongation initiation factor (LeIF) is a virulence factor expressed in both stages of all species of Leishmania (Cordin et al. 2006), being involved in transcription, translation, RNA export and degradation, pre-mRNA splicing and ribosomal biogenesis (Fuller-Pace 1994). The present in silico study was aimed at identification of immunogenic epitopes of four antigenic compounds from L. infantum, i.e. histone H1, KMP11, LACK and LeIF, in order to assemble a novel multi-epitope vaccine candidate to be used against VL.
Materials and methods
Retrieval of protein sequences
The amino acid sequences of L. infantum histone H1 (Accession no. ABB22792), KMP11 (Accession no. CAC9544709.1), LACK (Accession no. ALP73427.1) and LeIF (Accession no. XP_001466106.1) were gathered in FASTA format from the National Center for Biotechnology Information (NCBI), available at http://www.ncbi.nlm.nih.gov (Sayers et al. 2021).
MHC-I binding epitope prediction and screening
In this study, 3 web servers were used to predict potential MHC-I binding peptides: Immune Epitope Database (IEDB), ProPred-I and SYFPEITHI. High affinity epitopes to mouse MHC-I alleles, including H2-Db, H2-Dd, H2-Kb, H2-Kd, H2-Kk and H2-Ld were recognized as immunodominant epitopes.
The ProPred-I server (http://crdd.osdd.net/raghava//propred1/) identifies MHC-I promiscuous regions in an antigen based on matrices for 47 MHC class-I alleles, with the highest accuracy of 75% at 4% threshold using default settings (Singh and Raghava 2003). In IEDB server a percentile rank is given to each epitope, having inverse correlation with affinity. Epitope prediction was done using 10-mer length and IEDB recommended method 2020.04 (NetMHCpan EL 4.0) (http://tools.iedb.org/mhci/) (Kim et al. 2012). Also, SYFPEITHI server was used to predict decamer peptides against the selected mouse MHC-I alleles (http://www.syfpeithi.de/) (Rammensee et al. 1999). At final step, potent overlapping peptides among three web servers were selected and assessed regarding antigenicity and allergenicity using VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) and AllerTOP v2.0 (https://www.ddg-pharmfac.net/AllerTOP/) servers, respectively. One epitope with highest antigenic index without allergenicity was ultimately selected from each antigen to be embedded in the final vaccine construct.
MHC-II binding epitope prediction and screening
Helper T lymphocyte (HTL) epitopes were predicted using IEDB, RANKPEP and MHCPred v2.0 servers, against 5 mouse MHC-II alleles, i.e. H2-IAb, H2-IAd, H2-IAs, H2-IEd and H2-IEb.
The MHC-II binding prediction tool of IEDB server, available at (http://tools.iedb.org/mhcii/), was utilized to predict 15-mer binders using NN-align 2.3 (NetMHCII 2.3) method (Kim et al. 2012). Another web server, RANKPEP (http://imed.med.ucm.es/Tools/rankpep.html), makes predictions of epitopes (9-mer) on the basis of position-specific scoring matrix (PSSM) with threshold percentage of 4% (Reche et al. 2002). MHCPred V.2.0 (http://www.ddg-pharmfac.net/mhcpred/MHCPred/) was also employed for prediction of MHC-II binding epitopes (Vakili et al. 2018). Finally, shared, high-ranked epitopes predicted by above servers were selected to evaluate their antigenicity and allergenicity using VaxiJen v2.0 and AllerTOP v2.0 servers. Only 1 antigenic, non-allergenic epitope was selected from each antigen to be included in the vaccine sequence.
Prediction and screening of cytotoxic T lymphocyte (CTL) epitopes
In order to predict CTL epitopes, the CTLpred server (http://crdd.osdd.net/raghava/ctlpred/) with combined method was employed, applying artificial neural network (ANN) and support vector machine (SVM) algorithms (Bhasin and Raghava 2007). Furthermore, defined epitopes were screened in terms of antigenicity, allergenicity and toxicity using VaxiJen v2.0, AllerTOP v2.0 and ToxinPred (http://crdd.osdd.net/raghava/toxinpred/) servers, respectively. For each antigen, only 1 peptide having highest antigenic score and without allergenic and toxic traits was selected for the vaccine model construction.
Design and assemblage of the multi-epitope vaccine model
The final multi-epitope vaccine model was engineered and assembled using the qualified MHC-binders and CTL epitopes for each candidate antigen. Hence, 12 immunogenic epitopes were accurately predicted in silico and screened using different web servers. These epitopic regions were linked together with separate linkers or spacers. The CTL and MHC-I epitopes were connected by “AAY” linker, while HTL epitopes were linked by GPGPG spacers. Moreover, the vaccine immunogenicity was enhanced by addition of a toll-like receptor 4 (TLR-4) agonist peptide (RS-09; sequence: APPHALS) to the N-terminal of the vaccine sequence with the aid of EAAAK linker. Notably, a 6 × histidine (6 × His) residue was considered at the C-terminal of the designed sequence for further protein purification purpose.
Evaluation of allergenicity, antigenicity, solubility and physico-chemical properties
Two web servers evaluated the allergenicity of the designed vaccine model: AllerTOP v2.0 (accuracy: 85.3%) and AllergenFP v1.0 (accuracy: 88%). The AllerTOP server (https://www.ddg-pharmfac.net/AllerTOP/) transforms protein sequences into the integral equal-length vectors based on auto cross covariance (ACC) (Dimitrov et al. 2014a). On the other hand, AllergenFP server (https://ddg-pharmfac.net/AllergenFP/) predicts allergens by a new alignment-free, descriptor-based fingerprint approach based on structural and physico-chemical characteristics (Dimitrov et al. 2014b).
The protein antigenicity was predicted using two servers: VaxiJen v2.0 (accuracy: 70–89%) and ANTIGENpro (accuracy: 82%). The VaxiJen server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) makes predictions based on ACC transformation of the submitted sequence into uniform vectors, which is truly relied on the chemical nature of proteins (Doytchinova and Flower 2007). Also, ANTIGENpro (http://scratch.proteomics.ics.uci.edu/) is a pathogen-independent, alignment-free tool of SCRATCH protein predictor server (Magnan et al. 2010).
ProtParam server was used for physico-chemical prediction, being available at https://web.expasy.org/protparam/. This server computes several parameters comprising molecular weight (MW), theoretical isoelectric pH (pI), amino acid composition, in vitro and in vivo protein half-life, aliphatic index, instability index and the grand average of hydropathicity (GRAVY) score (Gasteiger et al. 2005). The Protein-Sol server (https://protein-sol.manchester.ac.uk/) was also employed for the prediction of protein solubility. Protein scores over 0.45 are considered as highly soluble (Hebditch et al. 2017).
Secondary and tertiary structure prediction
The Self-Optimized Prediction Method with Alignment (SOPMA) (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) is a web server to predict the secondary structure of a given protein (Geourjon and Deleage 1995). Next, the LOMETS (LOcal MEta-Threading-Server, version 3) web server (https://zhanglab.ccmb.med.umich.edu/LOMETS/) was used for homology modelling with full-length atomic models constructed by template-based fragment assembly simulations based on the results from ten threading programs, including FUGUE, HHsearch, MUSTER, PPA, PROSPECT2, SAM-T02, SPARKSX, SP3, FFAS, and PRC (Wu and Zhang 2007).
Refinement of the 3D model and validations
Refinement was done using GalaxyRefine server, available at http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE. For this purpose, the high-ranked 3D model predicted by LOMETS was submitted to GalaxyRefine server for further rehashing and mild/aggressive quality improvement. The output is provided by several parameters, including global distance test-high accuracy (GDT-HA), root mean square deviation (RMSD), MolProbity, Clash score, Poor rotamers and Rama favored (Heo et al. 2013). Subsequently, three web servers validated the refinement process: ERRAT (https://saves.mbi.ucla.edu/) (Colovos and Yeates 1993), Prosa-Web (https://prosa.services.came.sbg.ac.at/prosa.php) (Wiederstein and Sippl 2007) and MolProbity (http://molprobity.biochem.duke.edu/) (Majid and Andleeb 2019).
Conformational B cell epitope prediction
ElliPro of the IEDB server (http://tools.iedb.org/ellipro/) is one of the well-known online tools for conformational B cell epitope prediction, which employs a significant AUC score of 0.732, using default options of 6 Å max distance and 0.5-min score. The prediction of epitopes occurs in a three-step process: calculation of neighbor residue clustering, residual protrusion index (PI) and protein shape. Of note, high-score resides may be associated with improved solvent accessibility (Ponomarenko et al. 2008).
Vaccine protein disulfide engineering
The geometric conformation and total stability of a protein could be improved by disulfide bond formation. Hence, DbD2 server (http://cptweb.cpt.wayne.edu/DbD2/index.php) was used to evaluate the possibility of the occurrence of such bonds in the chimeric protein, via making cysteine mutations in those residues located at the highly-mobile area of the vaccine sequence (Craig and Dombkowski 2013).
Molecular interaction between the vaccine model and TLR-4 ligand
At first, the 3D structure of the receptor, TLR4/MD2 (Accession no. 3FXI) was retrieved from the PDB database of Research Collaboratory for Structural Bioinformatics (RCSB) (https://www.rcsb.org). ClusPro 2.0 protein-protein docking server was used to predict the binding affinity between the designed vaccine model and the examined receptor with default settings (Kozakov et al. 2010). The output of the server is provided as a top-rank cluster, among which the best docking pose is selected for visualization.
Codon optimization and in silico cloning
High-level protein expression in E. coli is a vital step for subunit vaccine production. Thus, reverse translation and codon optimization were performed by reverse translate tool of Sequence Manipulation Suite (https://www.bioinformatics.org/sms2/rev_trans.html) and JCat server (http://www.jcat.de/), respectively. Several key features of a DNA sequence such as codon adaptation index (CAI) and GC content are appraised using JCat, which play a major role in efficient expression of a chimeric protein in the respective host. In this section, codon optimization was done for better expression in E. coli K12 strain. At final stage, the suitable restriction sites of Eco53KI and EcoRV restriction enzymes were added to the 5′ and 3′-OH of the codon adapted vaccine sequence.
Immune simulation
The virtual immune simulation profile provoked by the finally approved vaccine model was predicted using C-ImmSim online server, available at http://150.146.2.1/C-IMMSIM/index.php. These predictions are premised on PSSM for machine learning methods. The output implies to three stimulated regions including bone marrow, thymus and lymph node (Rapin et al. 2010). This computer-aided simulation was accomplished using default parameters with random seed 12,345, simulation volume 10 and simulation steps 100. The output was compared to simulation data from A2 protein of L. chagasi (Accession No: GQ290460) as a representative immunogenic protein (Coelho et al. 2003).
Results
Prediction of MHC-binders and CTL epitopes
Highly antigenic, non-allergenic MHC-I binding epitopes (Supplementary File 1) and MHC-II binding peptides (Supplementary File 2) were selected for each examined L. infantum protein through a multi-step process using multiple web servers. Moreover, CTL epitopes with highest antigenic score and without allergenic and toxic features were included in the final vaccine construct (Supplementary File 3).
Engineering and assemblage of the multi-epitope peptide vaccine
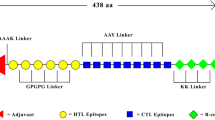
Based on the shared, high-ranked, antigenic CTL, HTL and MHC-I binding epitopes, totally 12 epitopic regions were selected from candidate proteins, which subsequently linked with appropriate linkers. Moreover, a TLR-4 agonist (RS-09 peptide) as a potent adjuvant and a histidine tag for purification were respectively added to the N- and C-terminal of the final vaccine sequence. Therefore, a 184 amino acid multi-epitope peptide vaccine was designed and assembled. A schematic representation of the vaccine design procedure, the employed web servers and the final vaccine construct are provided in Fig. 1.

Schematic representation of the multi-epitope vaccine design, used web servers and the final vaccine construct (184 residues). Those servers highlighted in green, red and purple were used for the prediction of MHC-I binding, MHC-II binding and cytotoxic T lymphocyte epitopes, respectively
Prediction of physico-chemical, solubility, antigenicity and allergenicity characteristics
The protein had an 18.99 kDa MW (immunogenic) with an alkaline theoretical pI (8.77). It was considered as stable (instability index: 34.91), relatively thermotolerant (aliphatic index: 66.20) and hydrophobic in nature (GRAVY score: 0.138). Other physico-chemical details of the vaccine model are provided in Table 1. Based on Protein-Sol server, the protein was soluble (0.562), while it was non-allergenic in nature based on AllerTOP and AllergenFP outputs. Furthermore, the probability of the whole vaccine antigenicity was calculated to be 0.7845 (threshold 0.5) by VaxiJen v2.0 server and 0.8546 by ANTIGENpro server, showing proper antigenic properties.
Secondary structure and homology modelling
SOPMA results for secondary structure prediction unleashed that 90 (48.91%) residues are alpha helix, 59 (32.07%) are random coil, 26 (14.13%) are extended strand and 9 (4.89%) are beta-turn (Fig. 2A). Ten 3D templates were generated by threading servers of LOMETS, among which 5a9q8 template had a good alignment based on normalized Z-score of 3.50. Also, the first model generated by the server was used for further refinement and validations (Fig. 2B).

(A) SOPMA server sequence-based (above) and graphical (below) secondary structure prediction, with Blue (Hh), orange (Cc), Red (Ee) and green (Tt) are alpha helix, random coil, extended strand and beta turn, respectively; (B) 3D model of the final vaccine construct shown as ribbon and surface predicted by LOMETS server
Refinement and validations of the tertiary modeled structure
The chosen 3D model was refined using GalaxyRefine server. This server introduced five rehashed models among which model number two was the best one, having GDT-HA of 0.9688, RMSD of 0.383, MolProbity of 2.371, Clash score of 21.5, Poor rotamers of 0.0 and Rama favored of 90.1. Based on Prosa-Web and ERRAT outputs, the quality of the model was shown to be improved from −5.93 Z-score and 70.45 quality factor in crude model to −6.02 Z-score and 75.00 quality factor in refined model. Moreover, MolProbity server result demonstrated that 80.2% of residues were in favored regions, while 96.7% of residues were located in allowed areas. After refinement, 90.1% and 98.4% of all residues were allocated to the favored and allowed regions, respectively (Fig. 3).

Confirmation of the quality of refined model provided by the GalaxyRefine server. (A) Based on MolProbity results for crude vaccine model, 80.2% of residues were in favored regions, while 96.7% of residues were located in allowed areas. (B) The overall crude model quality by Prosa-Web server was a Z-score of −5.93. (C) After refinement, 90.1% and 98.4% of all residues were allocated to the favored and allowed regions, respectively, as illustrated in MolProbity server. (D) The refined vaccine model showed improvement in quality as demonstrated by a Prosa-Web Z-score of −6.02
Prediction of conformational B cell epitopes
Based on ElliPro tool of the IEDB server, six conformational B cell epitopes were predicted in the refined vaccine sequence with the following scores: 1) 26 residues (0.834), 2) 16 residues (0.736), 3) 12 residues (0.669), 4) 6 residues (0.645), 5) 19 residues (0.587) and 6) 27 residues (0.559) (Fig. 4).

Predicted conformational B cell epitopes of the chimeric vaccine by ElliPro tool of IEDB analysis Resource. Length and score of each epitope are given in parentheses
Vaccine protein disulfide engineering
In total, DbD2 server showed that 24 residues possessed the potential to establish disulfide bonds. However, chi3 and B-factor energy (Kcal/mol) screening of the residues elucidated that only one residue pair (GLY 100 – ALA 160) could actually satisfy the formation of disulfide bonds, if mutated to cysteine. Screening of the residues was done on the basis of <2.5 energy value and − 87 to +97 chi3 value.
Molecular docking with TLR-4
The ClusPro 2.0 server was used for molecular docking procedure of the vaccine model with TLR4/MD2 receptor. First rank and most populated docking cluster (50 members) with the highest binding score (−940.6) was chosen for the visualization of the binding conformation between both examined molecules. As depicted in Fig. 5, the designed vaccine peptide has interactions with chains A (red) and C (yellow) of the receptor complex.

The binding conformation of the designed multi-epitope vaccine and TLR-4/MD-2 receptor complex. The designed peptide is in association with chains A (red) and C (yellow) of the receptor
Codon optimization and in silico cloning
Reverse translation of the protein sequence into the DNA sequence was done using the reverse translate tool of the Sequence Manipulation Suite. In the following, the sequence was submitted to the JCat server for codon optimization and improved expression level in E. coli K12 strain. The CAI value and GC% of the initially submitted sequence were 0.56 and 61.77, respectively, whereas these were improved in the codon optimized sequence as 1.0 and 55.07, correspondingly. These obtained results suggest that the expression of enhanced DNA sequence of the vaccine is maximum in the selected host.
Immune simulation profile
In silico evaluation of the immune response profile of the vaccine candidate revealed the generation of appropriate immunity against L. infantum infection (Fig. 6). In comparison with L. chagasi A2 protein, our vaccine model showed elevated active and presenting macrophage population (about 120 cells/mm3), progressively high levels of helper T memory cells (over 5500 cells/mm3 for up to 30 days) and relatively higher IFN-γ levels (over 400,000 ng/ml). Regarding A2 protein immune profile, no helper T memory cells were observed, while cytotoxic T cells showed a more rapid upsurge than our vaccine model but the persistence of such population was proportionately equal between the two proteins (Supplementary File 4).

The output of in silico immune simulation with the subunit vaccine provided by C-ImmSim server. (A) Level of cytokines (ng/ml) induced by the vaccine injection. The insert plot shows the IL-2 level with the Simpson index, D indicated by the dotted line, (B) T cytotoxic cell population per state (cells/mm3) upon antigen injection, (C) T helper cell population per state (cells/mm3) after antigen injection. (D) Macrophage population per state (cells/mm3) after antigen injection
Discussion
Vaccination is an outstanding preventive strategy for rapid, affordable and efficient improvement of the public health and infectious diseases control (Yaqub et al. 2014). Current subunit vaccines, with special emphasis on multi-epitope platforms, which harbor particular immunogenic components of a given pathogen, are more focused than whole organism and/or attenuated vaccines (Parvizpour et al. 2020). This approach is facilitated using bioinformatics and immunoinformatics web servers and/or standalone programs, which could efficiently discover the novel vaccine targets, i.e. immunogenic B- and T cell epitopes, from a huge bulk of genomic and proteomic information and direct them towards rational vaccine design (Goumari et al. 2020). In current study we exploited their immunodominant epitopes from histone H1, KMP11, LACK and LeIF proteins to develop a multi-epitope vaccine model using immunoinformatics web servers.
Since L. infantum is an intracellular parasite, Th-1 immune profile play a critical role to control the expansion of the infection, while neutralizing antibodies are less involved in protection. Accordingly, vaccine candidates composed of only specific T cell epitopes are more in focus (Joshi et al. 2014); thus in the present study we discovered, screened and selected only T cell epitopes from the above-mentioned protein targets (Histone H1, KMP11, LACK and LeIF). In details, HTL epitopes were predicted against various alleles of mouse MHC-I (H2-Db, H2-Dd, H2-Kb, H2-Kd, H2-Kk and H2-Ld) and MHC-II (H2-IAb, H2-IAd, H2-IAs, H2-IEd and H2-IEb) molecules, using cross-validating approach via multiple bioinformatics servers. Next, shared epitopes were selected and screened regarding antigenicity and allergenicity. As well, CTL-specific epitopic regions of each examined antigen were predicted and screened in terms of antigenicity, allergenicity and toxicity. Ultimately, high-scored, antigenic, non-allergenic and non-toxic peptides were chosen to assemble a multi-epitope vaccine construct using specific linkers, i.e. “AAY” (CTL), “GPGPG” (HTL) and “EAAAK” (adjuvant). In general, linkers are crucial in producing flexible molecules (extended conformation), proper folding of proteins and separation of functional domains, hence rendering the protein structure more stable (Dong et al. 2020).
Adjuvants act as innate immune catalysts, which initiate focal inflammatory sites that is relieved by antigen-TLR interactions. In this sense, the aluminum-based adjuvants are mostly the first choice for the majority of vaccines, however, they represent various side effects (Shanmugam et al. 2012). TLRs are a group of pattern recognition receptors (PPRs), specialized for the detection of pathogen-associated molecular patterns (PAMPs) (Coffman et al. 2010; Suresh and Mosser 2013). Recently, novel synthetic peptides mimicking the interaction of bacterial lipopolysaccharide (LPS) with TLR-4 receptor, can substantially balance signal transduction pathways, likewise those triggered by the adjuvants. Hence, they could be recognized as LPS mimotopes having particular association with TLR-4 receptor, so they’re called TLR-4 agonists (Shanmugam et al. 2012). The potential low immunogenicity of the multi-epitope vaccine was obviated by means of adding a TLR-4 agonist peptide (RS-09; sequence: APPHALS), as a natural adjuvant, to the N-terminal of the designed sequence. Upon binding to TLR-4, this peptide activates dendritic cells (DCs), leading to the polarization of naïve T CD4+ and CD8+ cells and subsequent secretion of IFN-γ and IL-2. Such a Th-1 immune profile is absolutely essential for the parasite clearance; hence, immunization against VL using natural adjuvants would be more effective (Vakili et al. 2018). Of note, a 6 × His sequence was applied in the C-terminal of the designed vaccine model to improve the purification process of the respective vaccine protein.
Along with the ability to elicit strong cell-mediated immune responses, an appropriate vaccine candidate should, also, possess admissible physico-chemical features during production steps. Based on bioinformatics analysis, the MW of the vaccine protein was 18.99 kDa with pI of 8.77, showing that the molecule is a potent immunogen (> 5–10 kDa) and it is relatively alkaline in nature. Moreover, this protein was proven to be stable (instability index: 34.91), hydrophobic in nature (GRAVY score: 0.138) and relatively thermotolerant (aliphatic index: 66.20). Additionally, the multi-epitope vaccine was highly soluble, antigenic and non-allergenic, which could be expressed efficiently in E. coli (K12 strain) after codon optimization. In the following secondary structure prediction results by SOPMA server showed that there were 48.91% alpha helix, 32.07% random coil, 14.13% extended strand and 4.89% beta turn in the sequence. Also, the 3D structure was modelled by LOMETS server, which needed refinement to yield a high-quality tertiary structure for docking process. Hence, GalaxyRefine server was used, which provided five refined models, among which the model with GDT-HA of 0.9688, RMSD of 0.383, MolProbity of 2.371, Clash score of 21.5, Poor rotamers of 0.0 and Rama favored of 90.1 was selected. After refinement, also, three web servers (ERRAT, Prosa-Web and MolProbity) confirmed the quality of the rehashed model. In the following the interaction between the refined vaccine model as a ligand and TLR-4 as a receptor was performed using ClusPro 2.0 server. As mentioned above, a small peptide (RS-09) as the TLR-4 agonist was added to the N-terminal of the vaccine sequence, which interacts with this receptor. The interplay between TLR-4 and its ligands has not been clearly elucidated (Wang et al. 2016); for instance, some ligands such as LPS, fusion protein of Respiratory Syncytial Virus (RSV) and Chlamydia pneumonia heat shock protein 60 bind to the receptor via a co-receptor named myeloid differentiation factor-2 (MD-2) (Peri and Piazza 2012; Rallabhandi et al. 2006; Wang et al. 2016). An internal pocket containing hydrophobic and positively-charged residues is formed by MD-2 co-receptor, which adequately fits amphipathic and negatively-charged agnostic molecules such as LPS (Park and Lee 2013). The findings of the molecular docking process showed that chains A (red) and C (yellow) of the receptor complex are in association with the multimeric vaccine. Finally, the immune response profile of the engineered multi-epitope vaccine was simulated using C-ImmSim server and compared with that of L. chagasi A2 protein. Our multi-epitope vaccine showed some advantages, including potent memory development in helper T cell population, relatively higher IFN-γ and active/presenting macrophage populations. Regarding A2 protein, no helper T memory cell was elicited and only a rapid upsurge was observed in cytotoxic T cell population, in comparison to our vaccine model, which both persisted equally.
Previously, some studies employed immunoinformatics approach to design and evaluate multi-epitope vaccine candidates against Leishmania parasites. Hashemzadeh et al. (2020) selected B- and T cell epitopes from GP63, KMP-11 and HSP-70 proteins of L. infantum and designed a multi-epitope vaccine construct using GGGGS and GSGSGS linkers and RpfE and RpfBG G5 domain of Mycobacterium tuberculosis as adjuvant. In contrast to our study, only preliminary in-silico analyses were done for the 45.9 kDa candidate without secondary structure prediction, protein disulfide engineering, molecular docking and evaluation of immunological profile (Hashemzadeh et al. 2020). Another study by Rabienia et al. (2020) utilized B- and T cell as well as IFN-γ inducing epitopes of L. major KMP-11 and HASPB to engineer a multi-epitope vaccine candidate (27.17 kDa), adjoined by GDGDG linker and profilin as adjuvant. Docking results demonstrated that profilin adequately binds well to TLR11 and the vaccine/receptor interaction is stable (Rabienia et al. 2020). Ropón-Palacios et al. (2019) exerted a somehow different strategy to develop a highly-efficacious vaccine candidate possessing 4 conserved epitopes among important Leishmania spp. in Latin America, including L. braziliensis, L. mexicana, L. panamensis and L. guyanensis. They utilized AAY and GPGP linkers to connect epitopes and 50S ribosomal protein L7/L12 as adjuvant. The 32.5 kDa vaccine candidate stably coupled with TLR4/MD2 receptor complex with strong H bond and hydrophobic interaction (Ropón-Palacios et al. 2019). In our study, we performed immune simulation to predict the immunological profile of the designed vaccine candidate upon injection, while such analysis was not performed in above studies. Moreover, α-helix was the most prominent secondary structure in our candidate, similar to the latter study, whereas random coils were most frequent in Rabienia et al. study. Notably, the MW of our vaccine candidate (about 19 kDa) was lower than that of previously-mentioned studies, which could be beneficial for future wet lab experiments such as purification purposes.
Conclusion
The gradual increase in the development of multi-epitope peptide vaccines is, to a great extent, due to their safety and rational design, which saves experimental resources and time. The present computer-aided study was aimed at accurate prediction of immunodominant CTL and HTL epitopes of four candidate proteins of L. infantum (Histone H1, KMP11, LACK and LeIF) to engineer and organize a multi-epitope-based subunit vaccine against VL. In total, the designed vaccine model showed acceptable structural, physico-chemical, antigenicity, allergenicity, solubility characteristics. As well, molecular docking with the TLR-4 receptor and the immune profile elicited by the vaccine candidate added more to the novelty and acceptability of this in silico designed molecular vaccine. However, in vitro and in vivo immunological studies are needed to confirm the efficacy of this maiden multi-epitope vaccine.
Availability of data and material
Data are available from the corresponding author on reasonable request.
Abbreviations
- CL:
-
Cutaneous Leishmaniasis
- VL:
-
Visceral Leishmaniasis
- MHC:
-
Major Histocompatibility Complex
- TCR:
-
T cell receptor
- APCs:
-
Antigen-presenting cells
- ROS:
-
Reactive oxygen species
- KMP11:
-
Kinetoplastid membrane protein 11
- LACK:
-
Leishmania-activated C-kinase
- LeIF:
-
Leishmania elongation initiation factor
- NCBI:
-
National Center for Biotechnology Information
- IEDB:
-
Immune Epitope Database
- HTL:
-
Helper T lymphocyte
- CTL:
-
Cytotoxic T lymphocyte
- ANN:
-
Artificial neural network
- SVM:
-
Support vector machine
- TLR-4:
-
Toll-like receptor-4
- ACC:
-
Auto cross covariance
- MW:
-
Molecular weight
- pI:
-
isoelectric pH
- GRAVY:
-
Grand average of hydropathicity
- SOPMA:
-
Self-Optimized Prediction Method with Alignment
- LOMETS:
-
Local meta-threading server
- GDT-HA:
-
Global distance test-high accuracy
- RMSD:
-
Root mean square deviation
- PI:
-
Protrusion index
- RCSB:
-
Research Collaboratory for Structural Bioinformatics
- CAI:
-
Codon adaptation index
- PPRs:
-
Pattern recognition receptors
- PAMPs:
-
Pathogen-associated molecular patterns
- LPS:
-
Lipopolysaccharide
- DC:
-
Dendritic cell
- RSV:
-
Respiratory Syncytial Virus
- MD-2:
-
Myeloid differentiation factor-2
References
Akhoundi M, Kuhls K, Cannet A, Votýpka J, Marty P, Delaunay P, Sereno D (2016) A historical overview of the classification, evolution, and dispersion of Leishmania parasites and sandflies. PLoS Negl Trop Dis 10:e0004349. https://doi.org/10.1371/journal.pntd.0004349
Bhasin M, Raghava G (2007) A hybrid approach for predicting promiscuous MHC class I restricted T cell epitopes. J Biosci 32:31–42. https://doi.org/10.1007/s12038-007-0004-5
Carmelo E, Martínez E, González AC, Piñero JE, Patarroyo ME, Del Castillo A, Valladares B (2002) Antigenicity of Leishmania braziliensis histone H1 during cutaneous leishmaniasis: localization of antigenic determinants. Clin Diagn Lab Immunol 9:808–811. https://doi.org/10.1128/CDLI.9.4.808-811.2002
Chakravarty J et al (2011) A clinical trial to evaluate the safety and immunogenicity of the LEISH-F1+ MPL-SE vaccine for use in the prevention of visceral leishmaniasis. Vaccine 29:3531–3537. https://doi.org/10.1016/j.vaccine.2011.02.096
Coelho EAF et al (2003) Immune responses induced by the Leishmania (Leishmania) donovani A2 antigen, but not by the LACK antigen, are protective against experimental Leishmania (Leishmania) amazonensis infection. Infect Immun 71:3988–3994. https://doi.org/10.1128/IAI.71.7.3988-3994.2003
Coffman RL, Sher A, Seder RA (2010) Vaccine adjuvants: putting innate immunity to work. Immunity 33:492–503. https://doi.org/10.1016/j.immuni.2010.10.002
Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2:1511–1519. https://doi.org/10.1002/pro.5560020916
Cordin O, Banroques J, Tanner NK, Linder P (2006) The DEAD-box protein family of RNA helicases. Gene 367:17–37. https://doi.org/10.1016/j.gene.2005.10.019
Craig DB, Dombkowski AA (2013) Disulfide by design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinf 14:1–7. https://doi.org/10.1186/1471-2105-14-346
Desjeux P (2004) Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis 27:305–318. https://doi.org/10.1016/j.cimid.2004.03.004
Desjeux P, Ghosh RS, Dhalaria P, Strub-Wourgaft N, Zijlstra EE (2013) Report of the post kala-azar dermal leishmaniasis (PKDL) consortium meeting, New Delhi, India, 27–29 June 2012. Springer
Dimitrov I, Bangov I, Flower DR, Doytchinova I (2014a) AllerTOP v. 2—a server for in silico prediction of allergens. J Mol Model 20:1–6. https://doi.org/10.1007/s00894-014-2278-5
Dimitrov I, Naneva L, Doytchinova I, Bangov I (2014b) AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30:846–851. https://doi.org/10.1093/bioinformatics/btt619
Djaoud Z, Parham P (2020) HLAs, TCRs, and KIRs, a triumvirate of human cell-mediated immunity. Annu Rev Biochem 89:717–739. https://doi.org/10.1146/annurev-biochem-011520-102754
Dong R, Chu Z, Yu F, Zha Y (2020) Contriving multi-epitope subunit of vaccine for COVID-19: immunoinformatics approaches. Front Immunol 11:1784. https://doi.org/10.3389/fimmu.2020.01784
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf 8:4. https://doi.org/10.1186/1471-2105-8-4
Freitas-Junior LH, Chatelain E, Kim HA, Siqueira-Neto JL (2012) Visceral leishmaniasis treatment: what do we have, what do we need and how to deliver it? Int J Parasitol Drugs Drug Resist 2:11–19. https://doi.org/10.1016/j.ijpddr.2012.01.003
Fuertes MA, Berberich C, Lozano RM, Gimenez-Gallego G, Alonso C (1999) Folding stability of the kinetoplastid membrane protein-11 (KMP-11) from Leishmania infantum. Eur J Biochem 260:559–567. https://doi.org/10.1046/j.1432-1327.1999.00217.x
Fuller-Pace FV (1994) RNA helicases: modulators of RNA structure. Trends Cell Biol 4:271–274. https://doi.org/10.1016/0962-8924(94)90210-0
Galanti N, Galindo M, Sabaj V, Espinoza I, Toro G (1998) Histone genes in trypanosomatids. Parasitol Today 14:64–70. https://doi.org/10.1016/s0169-4758(97)01162-9
Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook 571–607. https://doi.org/10.1385/1-59259-890-0:571
Geourjon C, Deleage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 11:681–684. https://doi.org/10.1093/bioinformatics/11.6.681
Goto Y et al (2011) KSAC, the first defined polyprotein vaccine candidate for visceral leishmaniasis. Clin Vaccine Immunol 18:1118–1124. https://doi.org/10.1128/CVI.05024-11
Goumari MM, Farhani I, Nezafat N, Mahmoodi S (2020) Multi-epitope vaccines (MEVs), as a novel strategy against infectious diseases. Curr Proteomics 17:354–364. https://doi.org/10.2174/1570164617666190919120140
Hashemzadeh P, Ghorbanzadeh V, Lashgarian HE, Kheirandish F, Dariushnejad H (2020) Harnessing Bioinformatic approaches to design novel multi-epitope subunit vaccine against Leishmania infantum. Int J Pept Res Ther 26:1417–1428. https://doi.org/10.1007/s10989-019-09949-6
Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J (2017) Protein–sol: a web tool for predicting protein solubility from sequence. Bioinformatics 33:3098–3100. https://doi.org/10.1093/bioinformatics/btx345
Heo L, Park H, Seok C (2013) GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res 41:W384–W388. https://doi.org/10.1093/nar/gkt458
Iborra S, Solana JC, Requena JM, Soto M (2018) Vaccine candidates against leishmania under current research. Exp Rev Vaccines 17:323–334. https://doi.org/10.1080/14760584.2018.1459191
Jardim A, Funk V, Caprioli R, Olafson R (1995) Isolation and structural characterization of the Leishmania donovani kinetoplastid membrane protein-11, a major immunoreactive membrane glycoprotein. Biochem J 305:307–313. https://doi.org/10.1042/bj3050307
Joshi S, Rawat K, Yadav NK, Kumar V, Siddiqi MI, Dube A (2014) Visceral leishmaniasis: advancements in vaccine development via classical and molecular approaches. Front Immunol 5:380. https://doi.org/10.3389/fimmu.2014.00380
Khatoon N, Pandey RK, Prajapati VK (2017) Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep 7:1–12. https://doi.org/10.1038/s41598-017-08842-w
Kim Y et al (2012) Immune epitope database analysis resource. Nucleic Acids Res 40:W525–W530. https://doi.org/10.1093/nar/gks438
Kozakov D et al (2010) Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19. Proteins 78:3124–3130. https://doi.org/10.1002/prot.22835
Kumari S, Kumar A, Samant M, Singh N, Dube A (2008) Discovery of novel vaccine candidates and drug targets against visceral leishmaniasis using proteomics and transcriptomics. Curr Drug Targets 9:938–947. https://doi.org/10.2174/138945008786786091
Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, Baldi P (2010) High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 26:2936–2943. https://doi.org/10.1093/bioinformatics/btq551
Mahida N, Blythe M, NDavies M, Doytchinova IA, Flower DR (2015) Toward the computer-aided discovery and design of epitope ensemble vaccines. Post-Genom Drug Vaccine Dev 5:205
Majid M, Andleeb S (2019) Designing a multi-epitopic vaccine against the enterotoxigenic Bacteroides fragilis based on immunoinformatics approach. Sci Rep 9:1–15. https://doi.org/10.1038/s41598-019-55613-w
Maroli M, Feliciangeli M, Bichaud L, Charrel R, Gradoni L (2013) Phlebotomine sandflies and the spreading of leishmaniases and other diseases of public health concern. Med Vet Entomol 27:123–147. https://doi.org/10.1111/j.1365-2915.2012.01034.x
Matsumura M, Fremont DH, Peterson PA, Wilson IA (1992) Emerging principles for the recognition of peptide antigens by MHC class I molecules. Science 257:927–934. https://doi.org/10.1126/science.1323878
Meeting WECotCotL, Organization WH (2010) Control of the Leishmaniases: Report of a Meeting of the WHO Expert Committee on the Control of Leishmaniases, Geneva, 22–26 March 2010. vol 949. World Health Organization
Molano I et al (2003) A Leishmania infantum multi-component antigenic protein mixed with live BCG confers protection to dogs experimentally infected with L. infantum. Vet Immunol Immunopathol 92:1–13. https://doi.org/10.1016/s0165-2427(02)00315-x
Nagill R, Kaur S (2011) Vaccine candidates for leishmaniasis: a review. Int Immunopharmacol 11:1464–1488. https://doi.org/10.1016/j.intimp.2011.05.008
Noazin S et al (2008) First generation leishmaniasis vaccines: a review of field efficacy trials. Vaccine 26:6759–6767. https://doi.org/10.1016/j.vaccine.2008.09.085
Park BS, Lee J-O (2013) Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med 45:e66–e66. https://doi.org/10.1038/emm.2013.97
Parvizpour S, Pourseif MM, Razmara J, Rafi MA, Omidi Y (2020) Epitope-based vaccine design: a comprehensive overview of bioinformatics approaches. Drug Discov Today 25:1034–1042. https://doi.org/10.1016/j.drudis.2020.03.006
Peri F, Piazza M (2012) Therapeutic targeting of innate immunity with toll-like receptor 4 (TLR4) antagonists. Biotechnol Adv 30:251–260. https://doi.org/10.1016/j.biotechadv.2011.05.014
Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, Peters B (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinf 9:1–8. https://doi.org/10.1186/1471-2105-9-514
Rabienia M, Roudbari Z, Ghanbariasad A, Abdollahi A, Mohammadi E, Mortazavidehkordi N, Farjadfar A (2020) Exploring membrane proteins of Leishmania major to design a new multi-epitope vaccine using immunoinformatics approach. Eur J Pharm Sci 152:105423. https://doi.org/10.1016/j.ejps.2020.105423
Rafati S et al (2005) Protective vaccination against experimental canine visceral leishmaniasis using a combination of DNA and protein immunization with cysteine proteinases type I and II of L. infantum. Vaccine 23:3716–3725. https://doi.org/10.1016/j.vaccine.2005.02.009
Rallabhandi P et al (2006) Analysis of TLR4 polymorphic variants: new insights into TLR4/MD-2/CD14 stoichiometry, structure, and signaling. J Immunol 177:322–332. https://doi.org/10.4049/jimmunol.177.1.322
Rammensee H-G, Bachmann J, Emmerich NPN, Bachor OA, Stevanović S (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–219. https://doi.org/10.1007/s002510050595
Rapin N, Lund O, Bernaschi M, Castiglione F (2010) Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One 5:e9862. https://doi.org/10.1371/journal.pone.0009862
Ready PD (2014) Epidemiology of visceral leishmaniasis. Clin Epidemiol 6:147. https://doi.org/10.2147/CLEP.S44267
Reche PA, Glutting J-P, Reinherz EL (2002) Prediction of MHC class I binding peptides using profile motifs. Hum Immunol 63:701–709. https://doi.org/10.1016/s0198-8859(02)00432-9
Requena JM, Alonso C, Soto M (2000) Evolutionarily conserved proteins as prominent immunogens during Leishmania infections. Parasitol Today 16:246–250. https://doi.org/10.1016/s0169-4758(00)01651-3
Rodrigues V, Cordeiro-da-Silva A, Laforge M, Silvestre R, Estaquier J (2016) Regulation of immunity during visceral Leishmania infection. Parasit Vectors 9:1–13. https://doi.org/10.1186/s13071-016-1412-x
Ropón-Palacios G, Chenet-Zuta ME, Otazu K, Olivos-Ramirez GE, Camps I (2019) Novel multi-epitope protein containing conserved epitopes from different Leishmania species as potential vaccine candidate: integrated immunoinformatics and molecular dynamics approach. Comput Biol Chem 83:107157. https://doi.org/10.1016/j.compbiolchem.2019.107157
Sayers EW et al (2021) Database resources of the national center for biotechnology information. Nucleic Acids Res 49:D10. https://doi.org/10.1093/nar/gkx1095
Shanmugam A, Rajoria S, George AL, Mittelman A, Suriano R, Tiwari RK (2012) Synthetic toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PLoS One 7:e30839. https://doi.org/10.1371/journal.pone.0030839
Singh H, Raghava G (2003) ProPred1: prediction of promiscuous MHC class-I binding sites. Bioinformatics 19:1009–1014. https://doi.org/10.1093/bioinformatics/btg108
Singh B, Sundar S (2012) Leishmaniasis: vaccine candidates and perspectives. Vaccine 30:3834–3842. https://doi.org/10.1016/j.vaccine.2012.03.068
Solano-Gallego L et al (2017) Diagnostic challenges in the era of canine Leishmania infantum vaccines. Trends Parasitol 33:706–717. https://doi.org/10.1016/j.pt.2017.06.004
Suresh R, Mosser DM (2013) Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv Physiol Educ 37:284–291. https://doi.org/10.1152/advan.00058.2013
Vakili B, Eslami M, Hatam GR, Zare B, Erfani N, Nezafat N, Ghasemi Y (2018) Immunoinformatics-aided design of a potential multi-epitope peptide vaccine against Leishmania infantum. Int J Biol Macromol 120:1127–1139. https://doi.org/10.1016/j.ijbiomac.2018.08.125
Valero NNH, Uriarte M (2020) Environmental and socioeconomic risk factors associated with visceral and cutaneous leishmaniasis: a systematic review. Parasitol Res 119:365–384. https://doi.org/10.1007/s00436-019-06575-5
Wang Y et al (2016) TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc Natl Acad Sci 113:E884–E893. https://doi.org/10.1073/pnas.1525639113
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:W407–W410. https://doi.org/10.1093/nar/gkm290
Wu S, Zhang Y (2007) LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res 35:3375–3382. https://doi.org/10.1093/nar/gkm251
Yaqub O, Castle-Clarke S, Sevdalis N, Chataway J (2014) Attitudes to vaccination: a critical review. Soc Sci Med 112:1–11. https://doi.org/10.1016/j.socscimed.2014.04.018
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
M. Shams and S. A. Shariatzadeh conceived the study protocol; M. Shams, M. Fatollahzadeh and H. Majidiani performed the bioinformatics analyses; A. Asghari assisted to prepare the revised version and improved the discussion; H. Irannejad performed the molecular docking process; M. Shams and H. Nourmohammadi drafted the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare none.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary File 1
Prediction, cross-validation and screening of MHC-I binding epitopes for L. infantum proteins (Histone H1, KMP11, LACK and LeIF). (DOCX 26 kb)
Supplementary File 2
Prediction, cross-validation and screening of MHC-II binding epitopes for L. infantum proteins (Histone H1, KMP11, LACK and LeIF). (DOCX 25 kb)
Supplementary File 3
Prediction and screening of CTL epitopes of L. infantum proteins (Histone H1, KMP11, LACK and LeIF). (DOCX 15 kb)
Supplementary File 4
Immune simulation results of the A2 protein of Leishmania chagasi. (DOCX 179 kb)
Rights and permissions
About this article

Cite this article
Shams, M., Nourmohammadi, H., Majidiani, H. et al. Engineering a multi-epitope vaccine candidate against Leishmania infantum using comprehensive Immunoinformatics methods. Biologia 77, 277–289 (2022). https://doi.org/10.1007/s11756-021-00934-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11756-021-00934-3