
Abstract
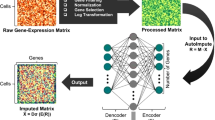
Single-cell RNA sequencing (scRNA-seq) technology has become an effective tool for high-throughout transcriptomic study, which circumvents the averaging artifacts corresponding to bulk RNA-seq technology, yielding new perspectives on the cellular diversity of potential superficially homogeneous populations. Although various sequencing techniques have decreased the amplification bias and improved capture efficiency caused by the low amount of starting material, the technical noise and biological variation are inevitably introduced into experimental process, resulting in high dropout events, which greatly hinder the downstream analysis. Considering the bimodal expression pattern and the right-skewed characteristic existed in normalized scRNA-seq data, we propose a customized autoencoder based on a two-part-generalized-gamma distribution (AE-TPGG) for scRNA-seq data analysis, which takes mixed discrete-continuous random variables of scRNA-seq data into account using a two-part model and utilizes the generalized gamma (GG) distribution, for fitting the positive and right-skewed continuous data. The adopted autoencoder enables AE-TPGG to captures the inherent relationship between genes. In addition to the ability of achieving low-dimensional representation, the AE-TPGG model also provides a denoised imputation according to statistical characteristic of gene expression. Results on real datasets demonstrate that our proposed model is competitive to current imputation methods and ameliorates a diverse set of typical scRNA-seq data analyses.
Article PDF
Similar content being viewed by others


Avoid common mistakes on your manuscript.
References
Potter S S. Single-cell RNA sequencing for the study of development, physiology and disease. Nature Reviews Nephrology, 2018, 14(8): 479–492
Li H, Courtois E T, Sengupta D, Tan Y, Chen K H, Goh J J L, Kong S L, Chua C, Hon L K, Tan W S, Wong M, Choi P J, Wee L J K, Hillmer A M, Tan I B, Robson P, Prabhakar S. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nature Genetics, 2017, 49(5): 708–718
Cao Y, Su B, Guo X, Sun W, Deng Y, Bao L, Zhu Q, Zhang X, Zheng Y, Geng C, Chai X, He R, Li X, Lv Q, Zhu H, Deng W, Xu Y, Wang Y, Qiao L, Tan Y, Song L, Wang G, Du X, Gao N, Liu J, Xiao J, Su X, Du Z, Feng Y, Qin C, Qin C, Jin R, Xie X S. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell, 2020, 182(1): 73–84.e16
Kharchenko P V, Silberstein L, Scadden D T. Bayesian approach to single-cell differential expression analysis. Nature Methods, 2014, 11(7): 740–742
Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek A K, Slichter C K, Miller H W, Mcelrath M J, Prlic M, Linsley P S, Gottardo R. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biology, 2015, 16(1): 278
Lun A T L, Bach K, Marioni J C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biology, 2016, 17(1): 75
Li W V, Li J J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nature Communications, 2018, 9(1): 997
Huang M, Wang J, Torre E, Dueck H, Shaffer S, Bonasio R, Murray J I, Raj A, Li M, Zhang N R. SAVER: gene expression recovery for single-cell RNA sequencing. Nature Methods, 2018, 15(7): 539–542
Van Dijk V, Sharma R, Nainys J, Yim K, Kathail P, Carr A J, Burdziak C, Moon K R, Chaffer C L, Pattabiraman D, Bierie B, Mazutis L, Wolf G, Krishnaswamy S, Pe’er D. Recovering gene interactions from single-cell data using data diffusion. Cell, 2018, 174(3): 716–729.e27
Basharat Z, Majeed S, Saleem H, Khan I A, Yasmin A. An overview of algorithms and associated applications for single cell RNA-seq data imputation. Current Genomics, 2021, 22(5): 319–327
LeCun Y, Bengio Y, Hinton G. Deep learning. Nature, 2015, 521(7553): 436–444
Bengio Y, Courville A, Vincent P. Representation learning: a review and new perspectives. IEEE Transactions on Pattern Analysis and Machine Intelligence, 2013, 35(8): 1798–1828
Hornik K. Approximation capabilities of multilayer feedforward networks. Neural Networks, 1991, 4(2): 251–257
Hinton G E, Salakhutdinov R R. Reducing the dimensionality of data with neural networks. Science, 2006, 313(5786): 504–507
Kadurin A, Nikolenko S, Khrabrov K, Aliper A, Zhavoronkov A. druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Molecular Pharmaceutics, 2017, 14(9): 3098–3104
Eraslan G, Simon L M, Mircea M, Mueller N S, Theis F J. Single-cell RNA-seq denoising using a deep count autoencoder. Nature Communications, 2019, 10(1): 390
Zhang Z, Cui F, Wang C, Zhao L, Zou Q. Goals and approaches for each processing step for single-cell RNA sequencing data. Briefings in Bioinformatics, 2021, 22(4): bbaa314
Mortazavi A, Williams B A, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nature Methods, 2008, 5(7): 621–628
Pickrell J K, Marioni J C, Pai A A, Degner J F, Engelhardt B E, Nkadori E, Veyrieras J B, Stephens M, Gilad Y, Pritchard J K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature, 2010, 464(7289): 768–772
Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-seq data. BMC Bioinformatics, 2011, 12(1): 480
Vallejos C A, Risso D, Scialdone A, Dudoit S, Marioni J C. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nature Methods, 2017, 14(6): 565–571
Li B, Ruotti V, Stewart R M, Thomson J A, Dewey C N. RNA-seq gene expression estimation with read mapping uncertainty. Bioinformatics, 2010, 26(4): 493–500
Belotti F, Deb P, Manning W G, Norton E C. Twopm: two-part models. The Stata Journal: Promoting communications on statistics and Stata, 2015, 15(1): 3–20
Lawless J F. Inference in the generalized gamma and log gamma distributions. Technometrics, 1980, 22(3): 409–419
Risso D, Perraudeau F, Gribkova S, Dudoit S, Vert J P. A general and flexible method for signal extraction from single-cell RNA-seq data. Nature Communications, 2018, 9(1): 284
Klein A M, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz D A, Kirschner M W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell, 2015, 161(5): 1187–1201
Minka T P. Estimating a gamma distribution. Microsoft Research, 2002, 1(3): 3–5
Chollet F. Keras. See Github.com/fchollet/keraswebsite
Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, et al. TensorFlow: large-scale machine learning on heterogeneous distributed systems. 2016, arXiv preprint arXiv: 1603.04467
Deng Q, Ramsköld D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science, 2014, 343(6167): 193–196
Kolodziejczyk A A, Kim J K, Tsang J C H, Ilicic T, Henriksson J, Natarajan K N, Tuck A C, Gao X, Bühler M, Liu P, Marioni J C, Teichmann S A. Single cell RNA-Sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell, 2015, 17(4): 471–485
Ding J, Adiconis X, Simmons S K, Kowalczyk M S, Hession C C, Marjanovic N D, Hughes T K, Wadsworth M H, Burks T, Nguyen L T, Kwon J Y H, Barak B, Ge W, Kedaigle A J, Carroll S, Li S, Hacohen N, Rozenblatt-Rosen O, Shalek A K, Villani A C, Regev A, Levin J Z. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nature Biotechnology, 2020, 38(6): 737–746
Arthur D, Vassilvitskii S. k-means++: the advantages of careful seeding. In: Proceedings of the 18th Annual ACM-SIAM Symposium on Discrete Algorithms. 2007, 1027–1035
Zheng G X Y, Terry J M, Belgrader P, Ryvkin P, Bent Z W, Wilson R, Ziraldo S B, Wheeler T D, McDermott G P, Zhu J, Gregory M T, Shuga J, Montesclaros L, Underwood J G, Masquelier D A, Nishimura S Y, Schnall-Levin M, Wyatt P W, Hindson C M, Bharadwaj R, Wong A, Ness K D, Beppu L W, Deeg H J, Mcfarland C, Loeb K R, Valente W J, Ericson N G, Stevens E A, Radich J P, Mikkelsen T S, Hindson B J, Bielas J H. Massively parallel digital transcriptional profiling of single cells. Nature Communications, 2017, 8(1): 14049
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay P K, Swerdlow H, Satija R, Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nature Methods, 2017, 14(9): 865–868
Xu C, Su Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics, 2015, 31(12): 1974–1980
Levine J, Simonds E, Bendall S, Davis K, Amir E A, Tadmor M, Litvin O, Fienberg H, Jager A, Zunder E, Finck R, Gedman A, Radtke I, Downing J, Pe’er D, Nolan G. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell, 2015, 162(1): 184–197
Francesconi M, Lehner B. The effects of genetic variation on gene expression dynamics during development. Nature, 2014, 505(7482): 208–211
Boeck M E, Huynh C, Gevirtzman L, Thompson O A, Wang G, Kasper D M, Reinke V, Hillier L W, Waterston R H. The time-resolved transcriptome of C. elegans. Genome Research, 2016, 26(10): 1441–1450
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant Nos. 62136004, 61802193), the National Key R&D Program of China (2018YFC2001600, 2018YFC2001602), the Natural Science Foundation of Jiangsu Province (BK20170934), and the Fundamental Research Funds for the Central Universities (NJ2020023). Thanks to all the open-minded researchers providing the codes and research resources. Thanks to all the anonymous reviewers.
Author information
Authors and Affiliations
Corresponding author
Additional information
Shuchang Zhao received the BSc degree from the Suzhou University, China in 2013, and MSc degree from the Anhui University of Science and Technology, China in 2016, respectively. Currently, he is working toward the PhD degree in the PARNEC group of the College of Computer Science and Technology at Nanjing University of Aeronautics and Astronautics (NUAA), China. His research interests include machine learning and bioinformatics.
Li Zhang received the BSc degree from the Changsha University of Science and Technology, China in 2007, and the MSc and PhD degrees from the Nanjing University of Aeronautics and Astronautics (NUAA), China in 2010 and 2015, respectively. He joined the College of Computer Science and Technology, Nanjing Forestry University, as a Lecturer, China in 2016. His current research interests include machine learning and bioinformatics.
Xuejun Liu received the BSc and MSc degrees in computer science from the Nanjing University of Aeronautics and Astronautics (NUAA), China in 1999 and 2002, respectively, and the PhD degree in computer science from the University of Manchester, UK in 2006. Currently, she is a professor in the PARNEC group of the College of Computer Science and Technology at NUAA, China. Her research interests include machine learning and its practical applications, including bioinformatics.
Electronic Supplementary Material
Rights and permissions
About this article

Cite this article
Zhao, S., Zhang, L. & Liu, X. AE-TPGG: a novel autoencoder-based approach for single-cell RNA-seq data imputation and dimensionality reduction. Front. Comput. Sci. 17, 173902 (2023). https://doi.org/10.1007/s11704-022-2011-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11704-022-2011-y