
Abstract
Thermodynamic optimization of the Fe-Mn-N system was performed using CALculation of PHADiagram method based on critical evaluation of all available phase diagrams and thermodynamic data. The Gibbs energies of liquid phase and solid solutions were described using the Modified Quasichemical Model and Compound Energy Formalism, respectively. The Gibbs energies of pure monoatomic N in both liquid and various solid states were newly reassessed for resolving the discrepancies left in the currently available database. Several existing problems in the previous assessments for the binary Fe-N and Mn-N systems were resolved and more precise and consistent description of the ternary Fe-Mn-N system was achieved with less number of model parameters, comparing to the previous assessment.
Similar content being viewed by others

1 Introduction
Nitrogen, as one of typical non-metallic impurity elements in steels, draws much attention among steelmakers because of non-negligible influence on the steel properties. In most cases, the impurity N is one of the undesirable elements in the steelmaking process. For example, formation of primary aluminum nitride (AlN) in high Al steels can result in serious defects like cracks. Moreover, the toughness and plasticity of steels deteriorate with the increase of dissolved N particularly at low temperatures. Therefore, N in low carbon steel typically needs to be tightly controlled during the steelmaking process. On the other hand, dissolved N in steels can also be beneficial to the stabilization of austenite, solid solution strengthening, and wear resistance improvement. Although the solubility of N in liquid and solid Fe is very low under one atmosphere pressure, it can be significantly increased with the presence of Mn, and is hard to be got rid of in high-Mn steels (15-25 wt.%Mn) and Mn alloys (74-90 wt.%Mn). As well known, because pure Mn and FeMn alloys are widely used in steelmaking process as Mn source, the refining of N in liquid Mn and FeMn is very important to cut down N source in steel production. In the Fe-Mn-N system, Fe and Mn nitrides can be formed under either N2 gas or NH3 gas in certain N2 partial pressure. These nitrides raise much interest because of their attractive physical properties like electronic and magnetic properties as well as potential applications to optical and wear-resistant coatings.[1,2]
To understand the thermodynamic behaviors of N in Fe, Mn, and Fe-Mn alloys, numerous experimental studies and computational modelings have been performed. Experimental studies on the subsystems of the Fe-Mn-N system were reviewed by Raghavan,[3,4,5] and Rokhlin and Lysova.[6] Based on available experimental data, Qiu[7] carried out thermodynamic assessments of the ternary Fe-Mn-N system using two-sublattice model for the solid solutions and Bragg–Williams Random Mixing Model (BWRMM) for the liquid phase. Four types of phases including liquid solution, solid solutions (α-CBCC based on α-Mn, β-CUBIC based β-Mn, γ-FCC, δ-BCC, and HCP), binary stoichiometric nitrides (γ′-Me4N, η-Mn6N4, and θ-Mn6N5) and N2 gas phase were taken into account in the description of the ternary Fe-Mn-N system. In their optimization, model parameters of the binary Fe-N,[8] Mn-N,[9] and Fe-Mn[10] systems were adopted from previous assessments. Unfortunately, the assessments of binary Fe-N and Mn-N systems adopted in the ternary systems have several critical problems. According to the thermodynamic parameters optimized by Frisk,[8] γ-FCC phase is calculated in high-N concentration region at the temperature below 470 °C, and this problem was still not resolved in the more recent reassessments by Du[11] and Gohring et al.[12] In addition, γ′-Mn4N phase of the Mn-N system was assumed as a stoichiometric compound in Qiu, and Fernandez and Guillermet’s[9] assessment, which is conflicting with experimental measurements by Hagg,[13] Brisi,[14] Juza et al.,[15] Lihl et al.,[16] Kudielka and Grabke,[17] Pompe[18,19] that indicate certain inhomogeneity range.
Thermodynamic database based on the CALculation of PHADiagram (CALPHAD) method is a powerful tool for new materials design and process optimization. Thermodynamic database is developed by means of thermodynamic modeling (optimization) of the target system, aiming at obtaining one set of the consistent Gibbs energies of all phases as functions of temperature and composition. During the optimization, all available thermodynamic and phase equilibrium data are critically and simultaneously evaluated. Thermodynamic property data (activity, entropy, and enthalpy, etc.) and phase diagram data can assist validation of each other for resolving the discrepancy among the available data. In this way, all the data are rendered self-consistent and complying thermodynamic principles. Moreover, prediction on unexplored thermodynamic properties and phase equilibria can be given by interpolations and extrapolations in a thermodynamically correct manner.
As part of the thermodynamic database of the new high alloy steel containing Fe-Mn-Al-Si-Cr-Ni-Ti-Nb-C-etc., thermodynamic optimization of the Fe-Mn-N system was performed in the present study based on critical evaluation of all available experimental data of the Fe-N, Mn-N, and Fe-Mn-N systems. The Modified Quasichmical Model (MQM)[20,21] and Compound Energy Formalism (CEF)[22] were used to describe the liquid and solid solutions, respectively. The parameters of all solid Fe-Mn phases by Huang[10] and those of liquid Fe-Mn solution by Paek[23] were adopted in the present study for the Fe-Mn system. The Gibbs energies of hypothetical liquid monoatomic N (\( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} \))[24] and each solid monoatomic N including \( g_{{{\text{N}}\left( {\text{BCC}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{FCC}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{CBCC}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{CUB}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{HCP}} \right)}}^{o} \) and \( g_{{{\text{N}}\left( {\gamma^{\prime } } \right)}}^{o} \) were newly assessed as part of the present optimization. All the thermodynamic calculations were performed using FactSage[25] software.
2 Thermodynamic Models
2.1 Pure Elements and Stoichiometric Compounds
The Gibbs energies of all pure elements were taken from Scientific Group Thermodata Europe (SGTE) database[26] and the Gibbs energies of all the stoichiometric compounds were determined based on available thermodynamic property data including heat capacity, standard enthalpy and entropy at 298.15 K. The Gibbs energies of pure elements and stoichiometric compounds can be calculated as follows:
where \( \Delta H_{298.15K}^{o} \) is standard enthalpy of formation from stable pure elements at 298.15 K, \( S_{298.15K}^{o} \) is the entropy at 298.15 K and \( C_{\text{P}} \) is the heat capacity. The heat capacity expression of each compound can be determined by fitting heat capacity data. If no reliable experimental data on \( C_{\text{P}} \) are available, \( C_{\text{P}} \) expression can be predicted using Neumann–Kopp (NK) rule.[27]
In the cases of elements or stoichiometric compounds exhibiting magnetic behavior, an additional magnetic contribution term \( \Delta G^{\text{mg}} \) needs to be accounted for describing the Gibbs energy of corresponding phase. \( \Delta G^{\text{mg}} \) is described by an empirical expression suggested by Inden[28] and modified by Hillert and Jarl[29] as follows:
where τ is given by \( T/T^{*} \) and T* is the critical temperature of magnetic transition associated with Curie temperature \( T_{\text{C}} \) for ferromagnetic materials or Neel temperature \( T_{\text{N}} \) for antiferromagnetic materials. β is the average magnetic moment per mole of atoms expressed in Bohr magnetons. \( g\left( \tau \right) \) is a polynomial fraction derived by Hillert and Jarl[29]:
where \( D = \frac{518}{1125} + \frac{11692}{15975}\left( {\frac{1}{P} - 1} \right) \), and the value of P can be considered as the fraction of the magnetic enthalpy absorbed above the critical temperature depending on the structure. P is 0.40 for the simple BCC_A2 phase while P is 0.28 for other common phases.
2.2 Liquid Solution
In the present study, the Modified Quasichemical Model (MQM)[20,21] accounting for the short-range ordering of the nearest-neighbor atoms was used to describe liquid solution. Compared to the conventional Bragg–Williams Random Mixing Model (BWRMM), the MQM gives more realistic description of the entropy of solution. In the MQM, the Gibbs energy of pair formation can be expanded as a polynomial in the pair fraction, instead of the component fraction, and the coordination numbers of components are allowed to vary with composition to more easily reproduce the short-range ordering in liquid solution. These provide greater flexibility in reproducing experimental data of the binary liquids and combining the binary parameters to higher order solutions.
In the case of the A − B binary liquid solution, the molecules of atoms A and B are distributed over the quasilattice sites. The atom pair exchanging reaction of the A − B liquid solution can be expressed as follows:
where (i − j) represents the nearest-neighbor pair between components i and j, and \( {{\Delta }}g_{AB} \) is the Gibbs energy change of forming 2 moles of (A − B) pairs. The Gibbs energy of the liquid solution can be calculated by the following equation:
where \( n_{A} \) and \( n_{B} \) are the numbers of moles of A and B atoms, and \( G_{A}^{o} \) and \( G_{B}^{o} \) are the molar Gibbs energies of pure A and B components. \( \Delta S^{{{\text{conf}} .}} \) is the configurational entropy of mixing given by random distribution of the (A − A), (B − B) and (A − B) pairs.
where \( n_{AA} \), \( n_{BB} \) and \( n_{AB} \) are the numbers of moles of (A − A), (B − B) and (A − B) pairs, and \( X_{AA} \), \( X_{AB} \) and \( X_{AB} \) are the pair fraction of the corresponding atom pairs. \( Y_{A} \) and \( Y_{B} \) are the coordination equivalent fractions of A and B components. The pair fractions \( X_{AA} \), \( X_{AB} \), \( X_{BB} \) and coordination equivalent fractions \( Y_{A} \), \( Y_{B} \) can be calculated as follows:
The \( \Delta g_{AB} \) is the model parameter for reproducing the Gibbs energy of the binary A − B solution, can be expanded as a polynomial in terms of the atomic pair fractions \( X_{AA} \) and \( X_{BB} \) as follows:
where \( \Delta g_{AB}^{o} \), \( g_{AB}^{i0} \) and \( g_{AB}^{0j} \) are the adjustable model parameters that can be functions of the temperature. In MQM, the coordination numbers of atoms A and B, \( Z_{A} \) and \( Z_{B} \), are allowed to vary with composition to reproduce the short-range ordering of the solution, and expand to higher-order systems using proper geometric interpolation technique:
where \( Z_{AA} \) is the value \( Z_{A} \) when all nearest neighbors of the A atom are A atoms, and \( Z_{AB} \) is the value of \( Z_{A} \) when all nearest neighbors are B atoms. \( Z_{BB} \) and \( Z_{BA} \) are defined in an analogous manner. In this study, \( Z_{ij} \) for all elements (i, j = Fe, Mn, or N) of the Fe-Mn-N system are set as 6, aiming at reproducing experimental data with a small number of model parameters.
The Gibbs energy of ternary liquid solution can be predicted using the interpolation technique based on the model parameters of binary systems. One of the advantages of MQM is the flexibility to choose the interpolation technique depending on the nature of each ternary system. For example, the binary Fe-N and Mn-N liquids show negative deviation from ideal solution, while liquid Fe-Mn solution is almost ideal solution. So Toop-type interpolation technique with N as the asymmetric component was used in the present study. Details of the interpolation technique and the Gibbs energy function of the ternary solution can be found in the reference by Pelton.[30]
2.3 Solid Solutions
The Gibbs energies of the solid solutions such as FCC, BCC, and HCP solutions were described using the Compound Energy Formalism (CEF)[22] considering their crystal structures. For example, when the solid solution is composed of two kinds of sublattices, (A, B)p (C, D)q (like substitutional and interstitial sites), its Gibbs energy of the solid solution per formula unit can be described using CEF as follows:
where \( y_{i} \) is the site fraction of species i in corresponding sublattice, \( G_{i:j} \) is the end-member Gibbs energy of \( \left( i \right)_{p} (j)_{q} \), p and q are the number of moles of related sublattice. \( L_{i, j:k} \) and \( L_{i:j,k} \) are adjustable interaction parameters between the species in the same sublattice, and \( G^{\text{mg}} \) is the magnetic Gibbs energy contribution. In the FCC, BCC, HCP, CBCC, and CUBIC solutions, Fe and Mn atoms occupy substitutional sites, while N occupies interstitial vacancy site. The non-stoichiometry of γ′ intermetallic phase is described by introducing the vacancy in N sites.
3 Critical Evaluation and Thermodynamic Optimization
Thermodynamic optimizations of the Fe-N, Mn-N and Fe-Mn-N systems were performed based on critical evaluation of all available thermodynamic and phase equilibria data. The binary Fe-Mn system was originally optimized by Huang.[10] Paek[23] recently reoptimized liquid phase using MQM. The model parameters of solid phases by Huang and liquid solution by Paek were accepted in the present study. The Gibbs free energy of liquid monoatomic N (\( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} \)) was recently reassessed by Paek et al.[24] to described the nitrogen dissolution behavior in liquid Fe-Mn-Al-Si-C solution. In particular, they proved that MQM can describe nitrogen solubility in the multicomponent liquid solution using very few model parameters. The descriptions of liquid Fe-N, Mn-N and Fe-Mn-N solutions by Paek et al.[24] were taken in this study without any modification. The Gibbs energies of various solid monoatomic N including \( g_{{{\text{N}}\left( {\text{FCC}} \right)}}^{o} \), \( g_{{N\left( {\text{BCC}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{CBCC}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{CUB}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{HCP}} \right)}}^{o} \) and \( g_{{{\text{N}}\left( {\gamma^{\prime}{\text{-FCC}}} \right)}}^{o} \) were assessed in the present study. The optimized Gibbs energies of various monoatomic N are summarized in Table 1. The Gibbs energies of Fe and Mn in gamma prime crystal structure are not available, so a small Gibbs energy difference (50 J/mol atom) between gamma prime Fe (or Mn) and FCC Fe (or Mn) value was assumed. The optimized thermodynamic model parameters of the binary Fe-N, Mn-N, Fe-Mn and ternary Fe-Mn-N systems are summarized in Table 2. The details of optimization are given below.
3.1 Assessments of the Gibbs Energies of Monoatomic N
3.1.1 Liquid Monoatomic N
The Gibbs energies of pure Fe and Mn in all states (solids, liquid, gas) are well known. However, pure monoatomic N in liquid and solid states are hypothetical, and their Gibbs energies are unavailable in the literature. In the description of Fe rich liquid solution, Wagner Interaction Parameter Formalism (WIPF)[31] is used widely. In this description, \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} \) = 1/2\( g_{{{\text{N}}_{2} \left( {\text{g}} \right)}}^{o} \) is assumed and the Henrian activity coefficient of N in liquid Fe solution is determined. In the previous assessment of the Fe-N system, Frisk[32] firstly proposed the Gibbs energy of pure liquid monoatomic N as \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} = 1/2g_{{{\text{N}}_{2} \left( {\text{g}} \right)}}^{o} - 711 + 70.2T \) to reproduce the Fe-N phase diagram. Later, Frisk[8] revised the original \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} \) to \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} = 1/2g_{{{\text{N}}_{2} \left( {\text{g}} \right)}}^{o} + 29950 + 59.02T \) because of difficulty to describe other N-containing systems such as the Cr-N and Mo-N. However, there is still a critical problem in the assessment of Fe-N system[32] related to \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} \). When N2 gas is suppressed, γ-FCC phase is calculated to reappears at high-N region (\( 0.4 < X_{\text{N}} < 0.5 \)) of the Fe-N system below 470 °C, which is ascribed to the relatively less stable Gibbs energy of pure liquid monoatomic N. The present authors[24] reassessed \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} \) by reproducing the solubility of N in liquid Fe. Because the Fe-N system is most well investigated among all liquid metal-N system, and the experimental solubility data of N in liquid Fe measured at widest temperature ranges among all available metal-N systems, we determined \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} \) by assuming liquid Fe-N solution is ideal solution. Compared to \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} = 1/2g_{{{\text{N}}_{2} \left( {\text{g}} \right)}}^{o} + 29950 + 59.02T \) by Frisk,[8] the presently obtained \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} = 1/2g_{{{\text{N}}_{2} \left( {\text{g}} \right)}}^{o} + 12643.9 + 45.939T \) is more stable in all temperature range.
3.1.2 Solid Monoatomic N
In the CALPHAD community, the Gibbs energy of diatomic N2 gas has been used to calculate the end-member Gibbs energies of the solid phases with additional Gibbs energy term. For example, the Gibbs energy of “MN” end-member of solid (F, M)1(N, Va)1 phase can be calculated by \( g_{{{\text{M}}:{\text{N}}}} = g_{{{\text{M}}\left( {\text{s}} \right)}}^{o} + 1/2g_{{{\text{N}}_{2} \left( {\text{g}} \right)}}^{o} \) + additional Gibbs energy with temperature dependence. Although this is a simple option to determine the Gibbs energy of end-member containing N, it may cause a large error in entropic term because the end-member Gibbs energy is determined to reproduce the experimental data mostly in low temperature range. This may cause unexpected stabilization of solid phase and difficulty in building a large and consistent thermodynamic database. Therefore, if possible, it would be better to reasonably define the Gibbs energy of hypothetical solid monoatomic N species.
In order to give more reliable and logical descriptions of solid phases, the Gibbs energies of various solid monoatomic N phases, including \( g_{{{\text{N}}\left( {\text{FCC}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{BCC}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{CBCC}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{CUB}} \right)}}^{o} \), \( g_{{{\text{N}}\left( {\text{HCP}} \right)}}^{o} \) and \( g_{{{\text{N}}\left( {\gamma^{\prime} {\text{-FCC}}} \right)}}^{o} \) were assessed in this work. Firstly, \( g_{{{\text{N}}\left( {\text{FCC}} \right)}}^{o} \) and \( g_{{{\text{N}}\left( {\text{BCC}} \right)}}^{o} \) were assessed based on the newly obtained Gibbs energy of pure liquid N (\( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} \)). According to the Richard’s rule,[33] the molar entropies of melting of FCC (9.6 J/mol K) and BCC (8.3 J/mol K) metals are constant. To apply this rule, the melting temperatures of both FCC and BCC monoatomic N were assumed to be 50 K based on the melting temperature of diatomic N2 (63.15 K). Hence, \( g_{{{\text{N}}\left( {\text{FCC}} \right)}}^{o} = g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} - 480 + 9.6T \) and \( g_{{{\text{N}}\left( {\text{BCC}} \right)}}^{o} = g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} - 415 + 8.3T \) were determined as Gibbs energies of pure monoatomic N in FCC and BCC crystal structures. The Gibbs energies of pure N in HCP (\( g_{{{\text{N}}\left( {\text{HCP}} \right)}}^{o} \)), CBCC (\( g_{{{\text{N}}\left( {\text{CBCC}} \right)}}^{o} \)), CUBIC (\( g_{{{\text{N}}\left( {\text{CUB}} \right)}}^{o} \)), and \( \gamma ^{\prime } {\text{-Me}}_{4} {\text{N}} \) (\( g_{{{\text{N}}\left( {\gamma^{\prime}} \right)}}^{o} \)) were assessed based on the above obtained \( g_{{{\text{N}}\left( {\text{FCC}} \right)}}^{o} \) and \( g_{{{\text{N}}\left( {\text{BCC}} \right)}}^{o} \) based on the assumption that the Gibbs energy differences of pure monoatomic N between different phases are the same as those of pure Fe. For example,\( g_{{{\text{N}}\left( {\text{HCP}} \right)}}^{o} - g_{{{\text{N}}\left( {\text{FCC}} \right)}}^{o} = g_{{{\text{Fe}}\left( {\text{HCP}} \right)}}^{o} - g_{{{\text{Fe}}\left( {\text{FCC}} \right)}}^{o} = - \;2243.4 + 4.31T \)].[25] The optimized Gibbs energies of liquid and solid monoatomic N species are summarized in Table 1.
3.2 Nitriding Treatment
In the nitriding experiments, N is mostly introduced into the sample using either diatomic N2 gas or NH3 gas. Gaseous N2 is basically used for nitriding when not very high N2 partial pressure is required for imparting desirable N amount into the sample, such as metallic Mn that has strong affinity to N. The nitriding reaction with N2 gas is expressed in Eq 19, and the dissolved N amount depends on the hydrostatic pressure of N2 gas and equilibrium reaction constant. When relatively high N2 partial pressure is required for nitriding treatment, NH3 gas is used at low temperature range (roughly below 700 °C) as an alternative nitriding gas which produces virtual N2 pressure by the NH3 dissociation reaction at the sample surface after adsorption process. H2 gas is mixed with NH3 gas to control N2 partial pressure, as indicated by nitriding reaction (20).
by combining two equations, partial pressure of N2 when NH3 gas is applied for nitriding can be calculated:
where \( P_{{{\text{NH}}_{3} }} \) and \( P_{{{\text{H}}_{2} }} \)are partial pressures of gaseous NH3 and H2 in the gas mixture, and Ke is equilibrium constant for the combined reactions of (19) and (20). In the present study, \( \Delta G^{o} \) was taken from FactPS database of FactSage software[25] and the calculated Ke is:
where T is the temperature in Kelvin.
3.3 The Binary Fe-N System
The Fe-N phase diagram was calculated by Hillert and Jarl[34] Agren,[35] and Kunze[36] based on available experimental data. Then, proper thermodynamic assessment of the entire Fe-N system was performed by Frisk[8,32] using CALPHAD method and later by Du,[11] Fernandez Guillermet and Du[37] and Gohring et al.[12] In the thermodynamic assessments, BWRMM was used for the liquid solution and two-sublattice model for the solid solutions. It is well known that Liquid, BCC (α-Fe and δ-Fe), FCC (γ-Fe), HCP (ε), γ′-Fe4N are stable in the binary Fe-N system. The overall review of the Fe-N system was given by Wriedt et al.[38]
3.3.1 Liquid Solution
In the Fe-N system, no experimental data of liquidus boundaries were available in the literature due to difficulty in carrying out experiments at high temperatures and N2 pressures. The solubility of N in liquid Fe under around 1 atm of N2 pressure was measured by many researchers using the Sieverts’ method,[39,40,41] the melts sampling method,[42,43,44,45] and the levitation melting method.[46,47,48,49] The experimental solubility of N2 gas in liquid Fe was plotted as a function of the inverse of the temperature in Fig. 1, along with the calculated solubility from the present authors[24] and Frisk.[8]

Variation of N solubility in liquid Fe with the inverse of the temperature
As shown in the figure, the experimental data are scattered heavily and the temperature dependences among various data are basically inconsistent. A number of experiments[16,40,44,45,48,50,51,52,53,54,55,56,57,58,59] were carried out to investigate the effect of N2 partial pressure on the solubility of N in liquid Fe over various temperatures from 1550 to 1900 °C. It was found that the solubility of N basically increases linearly with N2 partial pressure in the format of square root of N2 (\( P_{{{\text{N}}_{2} }}^{1/2} \)) in both low and high pressures regions, indicating the Henrian solution behavior of N dissolution in liquid Fe. However, the nitrogen solubility reported by Tsu and Saito[48] using levitation experiments at 1900 to 2500 °C shows decreasing trend above 2200 °C, and these can be well explained by considering N2 and Fe gas simultaneously as pointed out by Paek et al.[24] This decreasing trend is mostly ascribed to dilution of N2 gas by Fe vapor when the temperature increases to certain level. On the other hand, Frisk[8] did not consider this phenomenon, so rather different assessment results was obtained to reproduce all the scattered data as much as possible. The details of the evaluation of the experimental data and assessment are given in the previous reference.[24]
3.3.2 α/δ-Fe (BCC) and γ-Fe (FCC) Phases
Experimental studies[56,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74] were made to measure the solubility of N in α/δ-Fe and γ-Fe solid solutions under approximately 1 atm N2 partial pressure. The data were plotted in Fig. 2, together with calculated N solubility of the present study and that by Du.[11] Most researchers[60,61,64,65,67,69,70] carried out N solubility measurements in BCC (α/δ-Fe) and FCC (γ-Fe) solutions in equilibrium with 1 atm pressure of pure N2 gas or N2-H2/Ar gas mixture using conventional Kjeldahl method. Fast and Verrijp[62] measured the solubility of N2 in α-Fe phase from 500 to 900 °C using internal friction method. By means of Sieverts’ method, the solubility of N in α-Fe and γ-Fe solid solutions at various temperatures was investigated by Fountain and Chipman,[63] Milinskaya and Tomilin.[68] McLellan and Farraro[71]carried out experiments in the temperature range from 675 to 1440 °C, the solubility of N in α-Fe, δ-Fe and γ-Fe was determined under a total 1 atm pressure of 95%N2-5%H2 gas mixture using fusion extraction method. More recently, Kunze and Dresden[72] tried to get rid of the discrepancy of previous measurements through modifying the experimental method, in which hot extraction method was adopted to study the N2 dissolution in solid Fe under 99%N2-1%H2 gas mixture in a total pressure of 1 atm at 850, 1300 and 1450 °C. The most recent experiments were carried out by Feichtinger et al.[56] who adopted a newly developed technique (TGA) to investigate N2 dissolution in both liquid and solid Fe in the range between 860 and 1600 °C. In their experiments, the finely grained iron specimens were prepared as the starting materials and nitrided by 1 atm N2 partial pressure of N2-H2 mixture.

Solubility of N in α/δ(BCC)-Fe and γ(FCC)-Fe phases with the inverse of temperature under 1 atm of N2 pressure
As can be seen in Fig. 2, the solubility of N in γ-Fe decreases consistently with increasing temperature over the range from 912 to 1394 °C, and almost all experimental data are in good agreement with calculations in previous and present study. To reproduce this solubility, one interaction parameters between vacancy and N was introduced for FCC solution. On the other hand, the solubility of N in both α-Fe and δ-Fe increases with increasing temperature, and the solubility of N in α-Fe and δ-Fe lie on the same straight line, indicating that the enthalpy of N2 dissolution in low-temperature α-Fe phase is the same as that in high-temperature δ-Fe phase. Unfortunately, significant scattering can be observed among experimental data of α/δ-Fe phases. For example, the data of α-Fe phase by Fast and Verrijp,[62] Corney and Turkdogan,[61] Milinskaya and Tomilin,[68] and Zitter et al.[70] show remarkable deviation from those by Schenck et al.,[66] Grieveson and Turkdogan,[67] McLellan and Farraro,[71] and Kunze and Dresden[72] that are favored in this work. Compared to previous assessment results in dashed lines, the present results of N solubility in α-Fe and δ-Fe phase in solid line well reproduces the reliable experimental data. In the present study, the solubility of nitrogen in BCC phase (α- and δ-Fe) was described using one constant end-member parameter without any interaction parameters between vacancy and N. However, the end-member term with big temperature dependence was required in Du’s assessment to reproduce experimental data.
To investigate phase boundary of γ-Fe phase, Atkinson and Bodsworth[69] studied N dissolution in solid Fe specimen at 660, 720 and 810 °C. During their experiments, desirable chemistry NH3 + H2 gas mixture was passed through the starting materials for nitriding treatment. The partial pressure of nitrogen (\( P_{{{\text{N}}_{2} }}^{1/2} \)) was determined by the NH3/H2 ratio, as discussed in section 3.2. As shown in the Fig. 3(a), single BCC, BCC + FCC, single FCC and FCC + HCP two phase region are developed in sequence with increasing nitrogen pressure at this low temperature range. In the relatively higher temperature range (950–1100 °C), experiments on the N solubility in γ-Fe were carried out by Schenck et al.,[74] Naka et al.,[75] and Bashchenko et al.[76] using the same techniques as in Atkinson and Bodsworth’s experiments, but N2 gas was applied instead of NH3-H2 gas mixture. Experimental results were compared with calculations in Fig. 3(b). The experimental results of nitrogen solubility at high temperatures (Fig. 3b) are well reproduced in the present study. On the other hand, the results of Atkinson and Bodsworth[69] at 720 and 810 °C are not well reproduced while their results at 660 °C are well reproduced in the present study. In fact, the results of Atkinson and Bodsworth[69] at 720 and 810 °C may not be accurate because NH3 gas can dissociate into N2 and H2 by itself (that is, the initial NH3 and H2 ratio can be changed in the system and therefore, it is difficult to calculate the real N2 pressure) and N desorption from the sample happen when the temperature is above certain level, as explained by Somers et al.[73]

Solubility of nitrogen in γ-Fe depending on the partial pressure of N2 gas (\( P_{{{\text{N}}_{2} }}^{1/2} \)) at various temperatures. (a) low temperature range, and (b) high temperature range
Many experimental studies[61,62,69,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93] were conducted to determine phase boundaries of BCC (α) and FCC (γ) phases in the Fe-N system. Comparison between available experimental data with calculated results are presented in Fig. 4. As can be seen in Fig. 4(a), there is very slight difference between previous and present calculations, and modest scattering regarding the α phase boundary in equilibrium with γ′-Fe4N is shown among various experimental works. For example, the nitrogen content by Rawlings et al.,[81] Nacken et al.,[83] Imai et al.,[85] Pyzylecki[87] and Stein et al.[92] are slightly higher than other results and not favored in the present optimization. A maximum solubility of \( x_{\text{N}} = 0.004 \) in α-Fe at 594 °C is calculated in the present optimization by reproducing the experimental data by Paranjpe et al.,[79] Corney and Turkdogan[61] and Pictch et al.[82]

Phase diagram of the Fe-N system in Fe-rich region. (a) α phase region, and (b) γ phase region
In Fig. 4(b), the boundaries of γ (FCC) phase in equilibrium with α (BCC) and ε (HCP) phases are plotted in the temperature range between 594 and 1000 °C. Good consistency can be seen among most of experimental measurements except the data given by Jack[90] and Woehrle et al.[93] who recommended a relatively narrow γ phase region. In Jack’s experiments,[90] NH3-H2 gas mixture was passed through Fe powders at a fast flow rate to get rid of variation of nitrogen partial pressure resulted from NH3 disassociation. However, the difficulty in preparing homogeneous Fe-N austenite was declared by the author, due to phase transformation during cooling process. This can be the main reason of discrepancy originated from the experiments. Woehrle et al.[93] proposed lower N content by EPMA at the FCC/FCC + HCP and FCC/FCC + γ′-Fe4N phase boundaries without giving clear explanation.
As shown in Fig. 4(b), the previous assessment result by Du[11] shows steeper FCC phase boundary against HCP phase in comparison to the experimental data and the present study. This may be attributed to their Gibbs energy of end-member FeN of FCC phase. Such wrong slope of phase boundary can cause more serious error of FCC phase region with increasing temperature. Even in the later optimization by Gohring et al.,[12] the phase boundary of FCC solution against HCP phase is less reliable as shown in Fig. 4(b).
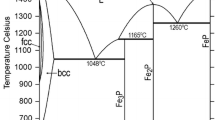
3.3.3 γ′-Fe4N Phase
In the present study, γ′-Fe4N phase was modeled as solid solution (Fe)4(N, Va)1 instead of a stoichiometric γ′-Fe4N compound. Grabke[94] investigated the N solubility in γ′-Fe4N at 450, 500, and 550 °C. The starting materials were nitrided with various NH3-H2 gas mixtures and the weight change was determined by gravimetrical method. Wriedt[95] carried out experiments on N dissolution in γ′-Fe4N phase using the same technique adopted by Grabke,[94] and reported the homogeneity range of N (\( x_{\text{N}} \)) between 0.1964 and 0.1994 at 500 °C, which is consistent with the results by Grabke. The above two sets of experimental results are shown in Fig. 5. In the temperature range from 450 to 550 °C, the solubility of N in γ′-Fe4N phase increases continuously with the increase of \( P_{{{\text{N}}_{2} }}^{1/2} \)until the N content (\( x_{\text{N}} \)) reaches a value of 0.1997 which is very close to stoichiometric N content of γ′-Fe4N. In order to reproduce these experimental data, we used both \( {}_{{}}^{o} L_{{{\text{Fe}}:{\text{N}},{\text{Va}}}}^{{}} \) and \( {}_{{}}^{1} L_{{{\text{Fe}}:{\text{N}},{\text{Va}}}}^{{}} \) interaction parameters. There are only a small difference among the previous assessment results by Du[11] and Gohring et al.[12] and the present work at 500 and 550 °C by Gohring et al., but the results at 450 °C by Gohring et al. are less consistent with experimental data by Grabke,[94] as can be seen in Fig. 5(a).

Variation of N solubility of γ′-Fe4N phase with \( P_{{{\text{N}}_{2} }}^{1/2} \) at temperatures of (a) 450, (b) 500, and (c) 550 °C
The phase diagram of γ′-Fe4N phase in equilibrium with FCC, BCC and HCP phases is presented in Fig. 6 along with experimental data. It is shown that experimental data are remarkably scattered particularly at temperatures between 450 and 550 °C. The results by Lehrer,[77] Grabke,[94] Wriedt,[95] Przylecki,[96] Woehrle et al.[93] are consistent with each other and accounted in the present optimization. The recent experiments were carried out by Van Voorthuysen et al.[97] using Elastic Recoil Detection (ERD) technique. In their experiments, the Fe-N phase diagram was extended down to 240 °C and a triple point of BCC, γ′-Fe4N and HCP phases (eutectoid reaction) at 214 ± 10 °C was induced, which was favored in the present optimization, as indicated in the figure. On the other hand, the assessments of Du[11] and Gohring et al.[12] calculated it at around 50 and 170 °C. The congruent dissociation point of γ′-Fe4N phase to HCP was calculated at 692 °C in both Goring et al.’s and present studies which are close to the experimental data, while the previous assessment by Du[11] calculates it at 702 °C. Besides, the eutectoid reaction of HCP → FCC + γ′-Fe4N is calculated at \( x_{\text{N}} = 0.162 \) at 648 °C in the present study, while it is calculated at \( x_{\text{N}} = 0.161 \) at 653 °C in Du’s[11] assessment and \( x_{\text{N}} = 0.164 \) at 652 °C in Gohring et al.’s assessment. Comparing to present optimization, the results by Goring et al. has more significant improvement in the boundary of HCP against γ′-Fe4N phase than the results by Du, but at the sacrifice of data fitting of γ′-Fe4N phase boundary as shown in the zoom area of the figure.

Phase diagram near γ′-Fe4N phase region
3.3.4 HCP Phase
The solubility of nitrogen in ε-Fe (HCP) phase was originally measured by Maldzinski et al.[98] using TGA technique from 350 to 725 °C. Because of high N content of HCP phase, NH3-H2 gas mixture in high nitrogen partial pressure was passed through for nitriding treatment. More recently, Somers et al.[73] carried out another set of experiments between 400 and 550 °C using the same technique as Maldzinski et al.[98] In both experiments, equivalent nitrogen partial pressure was controlled by adjusting the ratio of NH3 to H2. To investigate the N dissolution behavior in HCP phase, open porous structure specimens were prepared first for accelerating nitriding treatment with NH3-H2 gas mixture. The N dissolution amount in the specimens was analyzed using TGA technique. As mentioned by the researchers, the nitrided specimens at higher temperatures were weighed at a closed furnace by cooling down to a temperature between 400 and 500 °C, aiming at avoiding N desorption. In particular, Somers et al.[73] used the NH3-H2 gas mixture to produce virtually maximum 109 atm partial pressure of N2 gas at 450 °C. Figure 7 presents experimental data in comparison with calculated results. The experimental data at 350 and 600 °C are well reproduced in the present study, as shown in Fig. 7(a) and (c). However, as can be seen in Fig. 7(b), the experimental data for HCP at 450 °C can be explained well only for \( x_{\text{N}} < 0.292 \) where \( P_{{{\text{N}}_{2} }}^{1/2} < 1900 \) atm1/2, but two phase assemblage of HCP + Liquid is calculated instead of single HCP phase at \( P_{{{\text{N}}_{2} }}^{1/2} \) > 1900 atm1/2. If Liquid phase is suppressed, the solubility of N in HCP phase depending on \( P_{{{\text{N}}_{2} }}^{1/2} \) in Fig. 7(b) can be well explained even up to \( P_{{{\text{N}}_{2} }}^{1/2} \) = 35,000 atm1/2. If a small amount (less than 5 wt.%) of liquid phase existed in the sample (HCP + Liquid two phase assemblage), liquid phase could not be well detected by XRD and the apparent N content would still increase with \( P_{{{\text{N}}_{2} }}^{1/2} \) as presented by the experimental data in Fig. 7(b). Maldzinski et al.[98] also reported the existence of HCP solution of which composition is \( x_{\text{N}} = 0.20 \) at the temperatures of 680 and 690 °C, but this might suffer from the inaccuracy in temperature control. γ’-Fe4N should be stable at \( x_{\text{N}} = 0.20 \)and this temperature, according to Fig. 6.

Solubility of N in HCP phase depending on the partial pressure of nitrogen (\( P_{{{\text{N}}_{2} }}^{1/2} \)) at various temperatures: (a) 350 °C, (b) 450 °C and (c) 600 °C
3.3.5 Phase Diagram of the Fe-N System
Stability diagram of Fe depending on nitrogen partial pressure is calculated in Fig. 8 and compared with experimental data.[61,77,87,89,92,94,96,97,98,99,100,101] The equilibrium among BCC, FCC, HCP, γ′-Fe4N and Liquid phases are plotted against logarithmic N2 partial pressure (log\( P_{{{\text{N}}_{2} }} \)) below 1000 °C. All experiments were carried out using NH3-H2 gas mixture for nitriding treatment. The N2 partial pressure (\( P_{{{\text{N}}_{2} }} \)) at each temperature was converted from the ratio \( P_{{{\text{NH}}_{3} }} /P_{{{\text{H}}_{2} }}^{3/2} \), as explained in section 3.2. The early experimental data by Lehrer[77] at relatively higher temperatures and recent data by Van Voorthuysen et al.[97] at lower temperatures are scattered significantly. The stability diagram calculated by Goring et al.[12] is generally close to that calculated in the present study. The present one matches much better with experimental data specially at low temperatures. As pointed out above in Fig. 6, the stability range (low temperature limit) of γ′-Fe4N phase in the present study agrees well with experimental data while the calculated diagram by Du[11] overestimates its stability.

Stability diagram of the Fe-N system below 1000 °C
The phase diagram of the Fe-N system calculated from the present study is compared with that by Du[11] and Goring et al.[12] in Fig. 9. As shown in the figure, erroneous FCC phase in Fig. 9(b) appearing at \( 0.4 < x_{\text{N}} < 0.5 \) in the diagram given by Du and Goring et al. is avoided in the present Fe-N diagram. The eutectic reaction of γ′-Fe4N → BCC + HCP in the present study occurs at 207 °C based on recent experimental studies by Van Voorthuysen et al.[97] One of other noticeable differences between the present study and the previous assessment by Du[11] is homogeneity ranges of FCC and HCP solid solution. There are no definite experimental data to tell the boundaries of FCC and HCP solution, but as pointed out in Fig. 4 the present diagram is more consistent with existing experimental data. All invariant reactions of the Fe-N system optimized in the present study are summarized in Table 3.

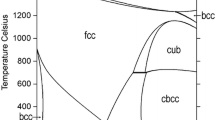
3.4 The Binary Mn-N System
It is well known that CBCC (α-Mn), CUBIC (β-Mn), BCC (δ), FCC (γ), HCP (ε), γ′-Mn4N, η-Mn3N2 (or Mn6N4) and θ-Mn6N5 and liquid phases are contained in the binary Mn-N system. According to the literature review, experimental information of the Mn-N system is incomplete. Most experimental studies were focused on the measurements of N solubility in liquid phase and solid FCC, HCP and γ′-Mn4N solutions. CBCC, CUBIC, FCC, BCC, HCP phases of the Mn-N system were widely accepted as solid solutions with N occupying the octahedral interstitial sites. However, there were some controversies among γ′-Mn4N, η-Mn3N2, and θ-Mn6N5 phases due to limited experimental information in the literature. In the thermodynamic assessment of the Mn-N system by Qiu and Fernandez Guillermet,[9] these three phases were modeled as stoichiometric compounds for simplifying the optimization, and the liquid phase was modeled using BWRMM by assuming random entropy mixing of atoms in the liquid, so excessive interaction parameters were used for reproducing experimental data. Because of these unrealistic assumptions, there were some discrepancies or inaccuracies that cannot be got rid of in describing N behavior in the binary Mn-N system and even higher-order systems. For example, the inhomogeneity range data within γ′-Mn4N phase cannot be explained based on previous modeling and this also results in inconsistency of description on the ternary γ′-(Fe, Mn)4N phase.
In the present study, the Mn-N system was thermodynamically optimized based on critical evaluation of all available thermodynamic and phase diagram data. The liquid solution was modeled using MQM accounting for short-range ordering. In addition, γ′-Mn4N and all other solid phases, except η-Mn3N2 and θ-Mn6N5, were modeled as solid solutions using CEF. Stoichiometry of η-Mn3N2 and θ-Mn6N5 phases were still adopted based on limited information including standard enthalpy of formation data and a few phase diagram datapoints. In the present optimization, more accurate descriptions on the thermodynamic properties and phase diagram were achieved with less model parameters comparing to the previous one.[9]
3.4.1 Liquid Phase
The solubility of N in liquid Mn was investigated by many researchers[48,102,103,104,105,106,107,108] mostly using melts sampling method and levitation melting method. Figure 10 presents the N solubility in liquid Mn under 1 atm N2 partial pressure calculated from the present authors[24] and previous assessment[9] along with experimental data. The N solubility increases with decreasing temperature. By adopting the \( g_{{{\text{N}}\left( {\text{l}} \right)}}^{o} \) proposed by Frisk,[8] Qiu and Fernandez Guillermet[9] modeled the liquid Mn-N solution using BWRMM by favoring the lower N solubility data by Beer,[103] Perepelkin,[105] Kor[107] based on melts sampling measurements. In contrast, the higher N solubility dataset by Tsu and Saito[48] and Kim et al.[108] were more favored in the present study. In order to improve the reliability of experimental data, Tsu and Saito[48] and Kim et al.[108] made corrections on the equilibrium N2 partial pressure by considering vaporization of Mn. More experimental investigation[48,103,105,106,107,108,109] on N solubility in liquid Mn were also carried out under various N2 partial pressures and temperatures. The decreasing solubility of nitrogen with increasing temperature was reproduced by introducing positive temperature dependent term of \( \Delta g_{\text{Mn N}}^{o} \). Details of thermodynamic optimization based on these measurements can be found in the assessment by Paek et al.[24]

Variation of N solubility in liquid Mn with temperature under 1 atm N2 pressure
3.4.2 The Solubility of N in FCC Phase
The solubility of N in FCC phase in equilibrium with various N2 partial pressure below 1 atm was measured by Kudielka and Grabke[17] and they are presented in Fig. 11. In the experiments, Mn powder was nitrided at 1025, 1075 and 1125 °C for about 4–5 h, followed by quenching. The N contents of quenched samples were analyzed using Kjeldahl method. It can be seen the solubility of N in FCC phase increases with the increase of N2 partial pressure (\( P_{{{\text{N}}_{2} }}^{1/2} \)), but decreases with the temperature. Compared to the previous assessment results by Qiu and Fernandez Guillermet,[9] the calculated results in the present study are more well consistent with experimental data.

Solubility of N in FCC Mn in equilibrium with N2 partial pressures
3.4.3 The Solubility of N in HCP and γ′-Mn4N Phases
In the experiments by Kudielka and Grabke,[17] the solubility of N in HCP phase was also measured at 900-1125 °C and less than 1 atm N2 partial pressure. Later, Pompe[18,19] investigated N2 dissolution in solid Mn using TGA, XRD and DTG techniques and reported HCP or γ′-Mn4N phase forming at 13-25 at.% of N. In Fig. 12, experimental data from the above studies are summarized in comparison with the present and previous calculations. As shown in Fig. 12(a), the solubility of N in HCP phase by Kudielka and Grabke[17] was well explained in the present study, but deviated from the calculated results by Qiu and Fernandez Guillermet.[9] Figure 12(b) shows the results of HCP and γ′-Mn4N phases by Pompe.[18,19] As γ′-Mn4N phase was treated as a stoichiometric compound in Qiu and Fernandez Guillermet’s assessment, their calculated results are not included in the figure. It should be noted that the disagreement related to HCP in Fig. 12(b) is due to the inconsistency between the results of Kudielka and Grabke[17] and Pompe.[18,19] Therefore, the accuracy of the results by Pompe[18,19] would be questionable.

The solubility of N in HCP and γ′-Mn4N phase under N2 partial pressure less than 1 atm. (a) HCP phase at 900 °C < T < 1125 °C and (b) γ′-Mn4N and HCP phase at 700 °C < T < 900 °C
Schenck et al.[66,74] applied a range of N2 partial pressure (\( P_{{{\text{N}}_{2} }} \)) from 1 atm to 64 atm to investigate the nitrogen dissolution behavior in Mn at 1000 °C. More recently, Ettmayer et al.[110] investigated the solubility of nitrogen in Mn at 1000 °C under \( P_{{{\text{N}}_{2} }} = 300\;{\text{atm}} \). The reported N solubility in HCP phase was compared with previous and present optimization results in Fig. 13. It shows that the data at relatively lower N partial pressures by Schenck et al.[66,74] were perfectly reproduced. However, the only datapoint at higher pressure by Ettmayer et al.[110] was slightly off calculated results indicating HCP + Mn6N5 two phase region. This may be due to the difficulty in detecting the trace amount of Mn6N5 mixed with HCP solid solution under the experimental conditions. According to the present calculation, only 0.7 at.% Mn6N5 compound is needed to be mixed with 99.3 at.% HCP solid solution to reproduce such data.

The solubility of N in HCP phase at 1000 °C under N2 partial pressure above 1 atm
3.4.4 η-Mn3N2 and θ-Mn6N5 Phases
According to the literature review, there was very limited experimental information regarding η-Mn3N2 and θ-Mn6N5. Although certain N solubility ranges of both η-Mn3N2 and θ-Mn6N5 were reported in the early literature,[13,14,16,111] there are still controversy among the available information. For example, η-Mn3N2 was reported to start decomposing at around 600 °C at 1 atm N2 partial pressure as suggested by Hagg[13] and Zwicker,[112] but was found to be stable up to 700 °C by Lihl et al.[16] and Mamporiya and Agladze.[113] Although several researchers reported the inhomogeneity range of η-Mn3N2 phase (Juza et al.[15]:\( 0.38 < x_{\text{N}} < 0.446 \) at 400 °C, Lihl et al.[16]:\( 0.382 < x_{\text{N}} < 0.41 \), and Brisi[14]:\( 0.35 < x_{\text{N}} < 0.355 \)), many researchers[114,115,116,117,118,119,120,121,122,123,124] reported the existence of stoichiometric compounds η-Mn3N2 and θ-Mn6N5. In the present study, therefore, η-Mn3N2 and θ-Mn6N5 were treated as stoichiometric compounds.
Tessier et al.[119] carried out drop solution calorimetry experiments on enthalpies of formation of Mn3N1.95 (η-Mn3N2 type) and Mn6N5.4 (θ-Mn6N5 type) from Mn(s) and N2(g). Recently, Yu et al.[124] indirectly investigated formation enthalpies of η-Mn3N2 using DFT calculation. Figure 14 presents the standard formation of enthalpy and entropy of intermediate compounds from α-Mn(s) and N2(g) in the Mn-N system. As shown in Fig. 14(a), the calorimetry measurements by Tessier et al.[119] are well reproduced within the experimental error range. The optimized standard formation entropy of η-Mn3N2 and θ-Mn6N5 are optimized to be − 31.472 and − 34.658 J/K/mol atoms, respectively, as shown in Fig. 14(b). The optimized thermodynamic values in the present study are close to the optimized results by Qiu and Fernandez Guillermet’s study,[9] except γ′-Mn4N.

Standard formation of enthalpy and entropy of intermediate compounds from α-Mn(s) and N2(g) in the Mn-N system
3.4.5 Phase Diagram of the Mn-N System
The optimized phase diagram of the Mn-N system is presented in Fig. 15 along with experimental data. All available experimental data[13,14,15,16,17,18,19,125,126] of phase boundaries and the solubility of N in various solid Mn were critically evaluated for determination of the Mn-N phase diagram. By comparing with previously assessed diagram,[9] the biggest change occurred in γ′-Mn4N phase that was modeled as solid solution in the present study instead of stoichiometric compound. Some other minor changes occurred in the CUBIC, FCC and HCP phases as well based on reported experimental results. The phase diagram with suppression of gas phase is also calculated in Fig. 15(b). In previously assessed diagram, hypothetical BCC phase is in equilibrium with Mn6N5 in the region of \( 0.45 < x_{\text{N}} < 0.82 \) near 0 °C, which is resolved in the present modeling. All invariant reactions of the binary Mn-N system in this work are summarized in Table 4.

In this study, the maximum solubility of N in α-Mn (CBCC) phase was determined at 0.48 at.%N at 605 °C, which is in agreement with the value (no more than 0.5 at.%N) suggested by Hagg.[13] and eutectoid FCC = CBCC + γ′-Mn4N reaction occurring at \( 605 \pm 3 \) °C proposed by Pompe.[18] Likewise, experimental measurements on β-Mn (CUBIC) phase are very scarce. The only available experimental information of CUBIC phase was from Juza et al.,[15] who carried out isothermal measurements of phase equilibria at 400, 600, 800 and 1000 °C. The CUBIC phase with 0.82 at.%N at 1000 °C obtained by means of XRD and Kjeldahl method. In addition, the specimens with 3.92 at.%N at 800 °C and 1.55 to 2.69 at.%N at 1000 °C were analyzed to be CUBIC + FCC two-phase solution. Pompe,[18] reported the eutectoid CUBIC = CBCC + FCC reaction at \( 645 \pm 5 \) °C. Kudielka and Grabke[17] investigated the Mn-N system over the range of 7.1 to 23.6 at.% N using XRD for the phase characterization and Kjeldahl method for the N analysis. The FCC, γ′-Mn4N and HCP phases from 660 to 1125 °C were studied systematically and eutectoid HCP = FCC + γ′-Mn4N reaction was found to occur at \( x_{\text{N}} = 0.13 \) and \( T = 800 \) °C, which was accepted in the present study. The experiments by Hagg,[13] Brisi,[14] Juza,[15] Lihl et al.,[16] Kudielka and Grabke,[17] and Pompe,[18,19] verified certain N solubility range in γ′-Mn4N phase at various temperatures, that is why γ′-Mn4N phase was modeled as solid solution instead of stoichiometric compound assumed in Qiu and Fernandez Guillermet’s assessment.[9] In the present study, the calculated boundary of HCP solution phase against γ′-Mn4N phase is more consistent with experimental data by Schenck et al.,[125] and Pompe[18] than previous assessment.[9] The decomposition temperatures of η-Mn3N2 and θ-Mn6N5 phase at 1 atm nitrogen pressure are optimized at 704 and 580 °C.
3.5 The Binary Fe-Mn system
The Fe-Mn system was originally optimized using a simple substitutional model by Kaufmann[127] and then revised by applying magnetic description to the BCC/FCC equilibrium by Huang.[128] Later, new experimental data[129] from the literature were taken into account in the re-optimization of the Fe-Mn system,[10] which has been widely used in most commercial thermodynamic databases. In the early twentyfirst century, Witusiewicz et al.[130,131] carried out experiment on enthalpy of formation and heat capacity of the Fe-Mn alloys using isoperibolic calorimetry technique, and the results were considered in the new thermodynamic modeling. Recently, Paek et al.[23] reoptimized the liquid phase of the binary Fe-Mn system using MQM and kept descriptions on the solid phases from Huang.[10] The updated Fe-Mn optimization by Paek et al.[23] was adopted in the present optimization of the Fe-Mn-N system. The calculated phase diagram of the Fe-Mn system is presented in Fig. 16.

Calculated phase diagram of the Fe-Mn system
3.6 Ternary Fe-Mn-N System
The ternary Fe-Mn-N system was thermodynamically optimized by Qiu.[7] Gas phase, liquid Fe-Mn-N solution, and eight solid phases including CBCC (α-Mn), CUBIC (β-Mn), FCC (γ), BCC (δ), HCP (ε), stoichiometric γ′-Me4N, η-Mn3N2 and θ-Mn6N5 compounds were considered in their optimization. Later, Kunze et al.[132,133] revised BCC parameters of the Fe-Mn-N system for reproducing their own experimental data of nitrogen solubility in Fe-Cr-Mn alloys. Though interaction parameters of the binary Mn-N system were avoided, excessive ternary parameters were still required in their optimization. As discussed above in the binary Mn-N system, γ′-Me4N phase was modeled as solid solution in the present optimization. The model parameters of binary Fe-N, Mn-N and Fe-Mn systems were accounted to perform thermodynamic optimization of the ternary Fe-Mn-N system. The liquid solution of the binary systems and ternary system were optimized by the present authors using MQM with proper interpolation technique and published previously.[24] The parameters of Huang[10] for the solid Fe-Mn system were adopted in the present optimization along with all other parameters described in sections 3.3 and 3.4. Experimental studies of the ternary system were mostly focused on liquid, FCC, HCP, BCC and γ′-Me4N phases, which will be explained in detail below.
3.6.1 Liquid Fe-Mn-N Solution
The solubility of N in Fe-Mn melts was investigated over wide temperature and composition ranges and various N2 partial pressures. Figure 17 presented the solubility of N in various Fe-Mn melts between 1550 and 1650 °C under different N2 pressures. Experimental data by many researchers[41,134,135,136,137] were plotted together with model calculation for comparison. As can be seen in Fig. 17(a), the N solubility increases with increasing Mn content, and the temperature dependence on N solubility shifts from positive to negative at 8.5 wt.%Mn under 1 atm N2 partial pressure. The early data by Wentrup and Reif[134] and Pehlke and Elliott[41] were negatively deviated from the calculations particularly at high Mn concentration region. The same discrepancies between the model calculation and measurements by other investigators[103,106,138] can also be observed in Fig. 17(b). This is probably due to enhanced interference of Mn vaporization on N2 partial pressure over the melts with increasing Mn content, which was unfortunately not taken into account in the early experiments. That is, due to the Mn vaporization at high Mn concentration melt, the actual partial pressure of N2 would not be maintained as originally designed in the experiment but lowered than original value. In recent experiments by Kim et al.,[108] Shin et al.[137] and Jang et al.,[139] the N solubility in Fe-Mn melts were determined using melts sampling method including correction of Mn vapor pressure. Apparently, the corrected data were in good agreement with model calculations throughout the considered ranges of Mn concentration and N2 partial pressure. The N behavior in Fe-Mn melts under various conditions was already described by the present authors[24] by comparing with experimental results.[41,50,55,102,103,106,108,109,134,135,136,137,138,139,140] More details can be found in the reference.[24]

Comparison between experimental data and calculations for N solubility in Fe-Mn melts of (a) Fe-rich side at 1550 to 1650 °C at \( P_{{{\text{N}}_{2} }} = 1 \) atm and (b) full-Mn range at 1550 °C at \( P_{{{\text{N}}_{2} }} = 0.2 \) and 1 atm
No ternary parameters for Fe-Mn-N liquid phase is required in the present study, while the previous study of Qiu.[7] introduced ternary parameters to reproduce the nitrogen solubility in Fe-Mn liquid phase.
3.6.2 FCC Solution
Several experimental studies[65,66,74,141] on the N solubility in FCC Fe-Mn alloys were made under various temperatures and N2 partial pressures. Mori et al.[65] measured N solubility in Fe-Mn alloys at 1050, 1150 and 1250 °C using Kjeldahl and Sieverts’ methods. In their experiments, the starting Fe-Mn alloys containing no more than 6.5 wt.% Mn were nitrided by 95%N2 + 5%H2 gas mixture of total 1 atm pressure. Grabke et al.[141] carried out experiments on N solubility in Fe-Mn alloys in equilibrium with 95%N2 + 5%H2 mixture between 900 and 1100 °C. The N2 partial pressure of 0.01 to 0.95 atm was passed through various Fe-Mn alloys containing 0.034 to 0.935 of Mn in mole fraction. In the experiments by Grabke et al.,[141] quenched samples containing less than 0.15 wt.%N were analyzed by means of hot extraction technique (LECO) and those containing above 0.15 wt.%N by conventional Kjeldahl method. The solubility of N in FCC Fe-Mn alloys is plotted in Fig. 18 as a function of Mn content from 900 to 1250 °C. As shown in Fig. 18(a), the solubility of N increases significantly with the increase of Mn content of the alloys. The present calculations are in good agreement with the experimental data by Mori et al.[65] In Fig. 18(b), the N solubilities at 900 and 1100 °C by Grabke et al.[141] are plotted against Mn concentration under 0.56 atm N2 partial pressure. Their data were well accounted throughout entire composition range of Fe-Mn. Overall, the nitrogen solubility in the present study is slightly higher than that from the previous assessment,[7] and becomes more consistent with experimental results.

Variations of N solubility in FCC Fe-Mn alloys with Mn contents from 900 °C to 1250 °C. (a) low Mn and (b) high Mn region
The temperature dependence of N solubility in Fe-rich FCC solution was also investigated in many early works.[60,66,70,142,143,144] All available experimental data are summarized in Fig. 19. Apparently, the solubility of N in FCC Fe-Mn alloys increases with decreasing temperature and increasing Mn content. The early data by Iwase and Fukusima[142] and Darken et al.[60] are lower than other experimental data at the same Mn content. Ko and Mclellan[144] measured N solubility in Fe-Mn alloys between 900 and 1250 °C by means of hot extraction method, and their data were considered not reliable due to inconsistency among the data. For example, the N solubility in Fe-8.471 wt.%Mn alloy is even slightly lower than that in Fe-6.894 wt.%Mn alloy, and there is almost no change of N solubility when Mn content increases from 9.853 wt.% increases to 14.97 wt.%, which is contradictory with all other results. In comparison, experimental measurements by Schenck et al.[66] are consistent with those by Kunze and Rothe,[143] which were favored in the present study.

Variation of N solubility in FCC Fe-Mn alloys with the temperature (\( P_{{{\text{N}}_{2} }} = 1 \) atm)
In the experiments by Schenck et al.[66] and Grabke et al.,[141] the effects of N2 partial pressure on N solubility in FCC Fe-Mn alloys were also investigated. Schenck et al.[66] investigated N2 dissolution in Fe-rich Fe-Mn alloys containing 2.9–9.5 wt.% of Mn at 1200 °C. The N2 partial pressure was controlled below 1 atm for nitriding treatment and the samples after equilibrium were quenched and followed by N analysis using conventional Kjeldahl method. Likewise, Grabke et al.[141] investigated the solubility of N in Fe-Mn alloys at 1000 °C under the N2 partial pressure below 1 atm as well. Various alloys containing 0.034 to 0.935 of Mn in mole fraction were used as starting materials. Comparison between the above experimental results and the present model calculations is presented in Fig. 20. The solubility of N increases almost linearly with the increase of N2 partial pressure (\( P_{{{\text{N}}_{2} }}^{1/2} \)) within 9.5 wt.%Mn in the alloys, and the present optimizations are in better agreement with the experimental data comparing to the previous one,[7] as shown in Fig. 20(a). In comparison, the solubility of N in high-Mn Fe-Mn alloys is plotted against the N pressure in the range of \( P_{{{\text{N}}_{2} }}^{1/2} < 1 \) atm1/2 in Fig. 20(b). As can be seen in the figure, the solubility of N in FCC solution increases continuously with increasing N2 partial pressure (\( P_{{{\text{N}}_{2} }}^{1/2} \)), but the dependence of N solubility on \( P_{{{\text{N}}_{2} }}^{1/2} \) becomes noticeably non-linear when the Mn content is more than 50.49 in weight percent. In addition, experimental data of the alloys containing more than 69.85 wt.%Mn are scattered heavily. This is probably due to N desorption from the samples during quenching treatment when the N2 pressure for keeping high N content was released.

Variations of N solubility in various FCC Fe-Mn alloys with N2 partial pressure (\( P_{{{\text{N}}_{2} }}^{1/2} < 1 \) atm1/2)
Schenck et al.[74] measured the solubility of N in various Fe-Mn alloys at 1000 °C in equilibrium with N2 pressure above 1 atm. In the experiments, a wider Mn range of Fe-Mn alloys containing 0.96–99.9 wt.% Mn were used as starting materials. As reported by Schenck et al.,[74] homogeneous FCC Fe-Mn solutions were obtained by nitriding Fe-Mn alloys containing less 55 wt.%Mn. Experimental N solubility data of high N2 partial pressure by Schenck et al.[74] and low N2 partial pressure by Grabke[141] are plotted against \( P_{{{\text{N}}_{2} }}^{1/2} \) in Fig. 21 along with model calculations. As shown in the figure, the solubility of N is proportional to N2 partial pressure as well as Mn concentration in the alloys. Apparently, experimental data at higher pressures are well accounted by the present optimization. As suggested by Schenck et al., Sieverts’ law is only obeyed when Mn content of the alloys is less than 3 wt.%.

Variations of N solubility in FCC Fe-Mn alloys with N2 partial pressure
In order to reproduce N solubility data in FCC solution in the wide temperature and composition range, it was necessary to introduce ternary interaction parameters, \( L_{{{\text{Fe}},\;{\text{Mn}}:{\text{N}}}}^{\text{FCC}} = 86609 - 66.107T - 23012\left( {y_{\text{Fe}} - y_{\text{Mn}} } \right) \) in the present study. Qiu also introduced similar ternary parameters in his assessment.[7]
3.6.3 N Dissolution in HCP Fe-Mn Alloys
Figure 22 shows the calculated nitrogen solubility in HCP solution along with the experimental data by Schenck et al.[74] Compared to the previous assessment by Qiu,[7] the calculated solubility in the present study is much more consistent with experimental data. No ternary parameters for HCP solution was introduced in the present study.

Variations of N solubility in FCC and HCP Fe-Mn alloys with N2 partial pressure (\( P_{{{\text{N}}_{2} }}^{1/2} > 1 \) atm1/2)
3.6.4 N Dissolution in BCC Fe-Mn Alloys
The solubility of N and precipitation of Mn nitrides in BCC Fe-Mn alloys were studied by Enrietto,[145] Nacken and Kuhlmann,[146] and Rawlings and Hatherley.[147] Enrietto[145] carried out experiments on N solubility in BCC Fe-Mn alloys containing 0.15-0.75 wt.% Mn between 250 and 500 °C. After nitriding Fe-Mn wire samples by reacting with anhydrous ammonia, Enrietto found the formation of γ′-Fe4N precipitates in Fe-Mn matrix and assumed all nitrogen in matrix and some Mn-associated nitrogen (called as “segregations with Mn” in the reference) as a part of soluble nitrogen in BCC phase. He used the internal friction method and wet chemical analysis to determine the N solubility. It seems that the size of Mn nitride precipitates is too small to distinguish under normal optical microscope. According to the phase diagram calculation shown in Fig. 23, γ′-Me4N (mainly γ′-Fe4N) and Mn6N5 phases can form simultaneously at the experimental condition of Enrietto.[145] In fact, segregation associated to Mn mentioned in his paper should be Mn6N5 phase. As Enrietto counted the nitrogen from Mn6N5 and solid solution, the experimental solubility should be higher than the calculated phase diagram as shown in Fig. 23. Therefore, the direct comparison of the experimental data with calculated diagram is impossible. But it should be noted that the N solubility measured by Enrietto[145] is almost the same as the solubility of N in BCC Fe with no Mn. Qiu[7] assumed that the nitrogen solubility by Enrietto[145] was obtained at \( P_{{{\text{N}}_{2} }} = 1 \) atm in his assessment, which is wrong.

Effects of Mn on the N solubility in BCC Fe-Mn solution. It should be noted that the solubility of N by Enrietto[145] contains both N in BCC solid solution and N from Mn6N5 phase
Figure 24 presented the solubility of N in BCC Fe-Mn solution at 585 °C. Two sets of experimental data were compared with the calculated results. The experimental N content was measured by Nacken and Kuhlmann[146] using the internal friction method and Rawlings and Hatherley[147] using the Kjeldahl technique. As can be seen in Fig. 24, both experimental results are very different from each other, and they are much higher than the calculated results. In both experimental analysis, the small Mn nitrides cannot be well detected as discussed above in Fig. 23. Therefore, the experimental data for the solubility in BCC Fe-Mn solution are less reliable. In order to reproduce such high N solubility in Fe-rich BCC solution, the standard Gibbs energies of Mn nitrides which are reasonably constrained in the binary Mn-N system should be changed significantly. In the present study, no ternary parameters are introduced in BCC solid solution.

The solubility of Mn nitrides in BCC Fe-Mn solution at 585 °C
3.6.5 Ternary CBCC and CUBIC Solid Solutions
CBCC and CUBIC phases are not stable on the binary Fe-N system. In the ternary Fe-Mn-N system, Mn atoms in the CBCC and CUBIC solutions can be partially substituted by Fe atoms. However, it is hard to determine Gibbs energies of end-member of FeN in both solid solution. In the present study, for example, we simply set \( G_{\text{Fe:N}}^{\text{CBCC}} = G_{{{\text{Fe}}\left( {\text{CBCC}} \right)}}^{o} + G_{{{\text{N}}\left( {\text{CBCC}} \right)}}^{o} + 200,000 \) J/mol to avoid any binary Fe-N interaction parameters and ternary parameters in Fe-Mn-N system. As results, the nitrogen solubilities in CBCC and CUBIC Mn are calculated to decrease significantly with increasing Fe content. No experimental data are available for the comparison.
3.6.6 Ternary γ′-Me4N Solution
The solubility of Fe in γ′-Mn4N in the temperature range between 500 and 900 °C was investigated by Burdese et al.[148] The γ′-Mn4N sample was prepared by nitriding the Fe-Mn samples with NH3 + H2 gas mixture, and the phase boundary of γ′-Me4N solution against Fe (FCC) was determined. The isothermal ternary Fe-Mn-N sections at 500, 600, 750 and 900 °C are calculated in Fig. 25 and compared with the experimental data. In order to reproduce the experimental boundary of γ′-Fe4N solubility in γ′-Mn4N, one ternary interaction parameter \( L_{{{\text{Fe}},\;{\text{Mn}}:{\text{N}}}}^{{\gamma^{\prime } }} = 19719 - 16.736T \) was introduced. Burdese et al.[148] reported the existence of γ′-Me4N solution at 900 °C, but it is calculated that HCP solid solution is stable. In the binary system, the transition temperature of γ′-Mn4N to HCP is about 882 °C, therefore the experimental data by Burdese et al.[148] might be measured slightly lower temperature than 900 °C.

Isothermal sections of the Fe-Mn-N system with the suppression of N2 gas at the temperatures of (a) 500 °C, (b)600 °C, (c) 750 °C, and (d) 900 °C
3.6.7 Predicted Isobaric Phase Diagram
According to the discussion above, thermodynamic database developed in the present study is more accurate in describing the behavior of N in the Fe-Mn-N system than previous studies. Figure 26 presents the predicted isobaric phase diagrams for the nitrogen dissolution in the Fe-Mn alloys at 1000 °C. The N2 partial pressure of 1, 10, 100 and 1000 atm were used in the calculations. As shown in the figure, in general, the phase transition of FCC → FCC + HCP → HCP occurs with increasing nitrogen pressure. At the same pressure, nitrogen solubility increases with Mn content because the affinity of Mn and N is stronger than that of Fe and N. Based on the present thermodynamic study, the isobaric phase diagram can be calculated at any temperature and composition. This information can be used for the development of high N steels.

Isobaric curve of nitrogen dissolution in the entire composition range of FeMn alloys at 1000 °C
4 Summary
Critical evaluation of available experimental data of the binary Fe-N, Mn-N and ternary Fe-Mn-N systems has been conducted to determine a set of Gibbs energies of all the phases involved in the Fe-Mn-N system. The thermodynamic behaviors of N in liquid Fe-Mn solution was described using Modified Quasichemical Model (MQM), and solid solutions such as BCC, FCC, CBCC, CUBIC and HCP etc. were modeled using two-sublattice Compound Energy Formalism (CEF). The present thermodynamic optimization resolved discrepancies in the available experimental data and previous assessments.
Change history
26 October 2018
In the originally published article, numbers in Eq 2 and 3 were transposed. The following are the full, correct equations:
References
H. Yang, R. Yang, A.R. Smith, and W.R. Lambrecht, Atomic-Scale Structure of η-Phase Mn3N2(0 1 0) Studied by Scanning Tunneling Microscopy and First-Principles Theory, Surf. Sci., 2004, 548(1–3), p 117–128
W.J. Feng, N.K. Sun, J. Du, Q. Zhang, X.G. Liu, Y.F. Deng, and Z.D. Zhang, Structural Evolution and Magnetic Properties of Mn-N Compounds, Solid State Commun., 2008, 148(5–6), p 199–202
V. Raghavan, The Fe-Mn-N (Iron-Manganese-Nitrogen) System, Trans. Indian Inst. Met., 1984, 37(3), p A–F
V. Raghavan, The Fe-Mn-N (Iron-Manganese-Nitrogen) System in Phase Diagrams of Ternary Iron Alloys, Ind. Inst. Met. Delhi, 1987, 1, p 183–189
V. Raghavan, Fe-Mn-N (Iron-Manganese-Nitrogen), J. Phase Equilib., 1993, 14(5), p 629
L. Rokhlin and L. Evgeniya, Iron-Manganese-Nitrogen, Iron Systems, Part 4, Springer, Berlin, 2008, p 197–214
C. Qiu, A Thermodynamic Evaluation of the Fe-Mn-N System, Metall. Trans. A, 1993, 24(3), p 629–645
K. Frisk, A Thermodynamic Evaluation of the Cr-N, Fe-N, Mo-N and Cr-Mo-N Systems, Calphad, 1991, 15(1), p 79–106
C. Qiu and A. Fernández Guillermet, Predicative Approach to the Entropy of Manganese Nitrides and Calculation of the Mn-N Phase Diagram, Z. Metalkd., 1993, 84(1), p 11–22
W. Huang, An Assessment of the Fe-Mn System, Calphad, 1989, 13(3), p 243-252
H. Du, A Reevaluation of the Fe-N and Fe-C-N Systems, J. Phase Equilib., 1993, 14(6), p 682–693
H. Göhring, O. Fabrichnaya, A. Leineweber, and E.J. Mittemeijer, Thermodynamics of the Fe-N and Fe-N-C Systems: the Fe-N and Fe-N-C Phase Diagrams Revisited, Metall. Mater. Trans. A, 2016, 47(12), p 6173–6186
G. Hagg, X-ray Investigation of the Nitrides of Manganese, Z. Phys. Chem., 1929, 4(B), p 346–370
C. Brisi, The System Manganese-Nitrogen, Metall. Ital., 1955, 47, p 405–408
R. Juza, H. Puff, and F. Wagenknecht, The System Manganese-Nitrogen, Z. Elektrochem. A. Phys. Chem., 1957, 61, p 804–809
F. Lihl, P. Ettmayer, and A. Kutzelnigg, The System Manganese-Nitrogen, Z. Metallkd., 1962, 53, p 715–719
H. Kudielka and H.J. Grabke, Investigation of the Mn-N Phase Diagram by X-Ray Diffraction at High Temperatures, Z. Metallkd., 1975, 66(8), p 469–471
R. Pompe, Investigation of the Phase Diagram Manganese-Nitrogen in the Range 10–25 at.% Nitrogen by Thermoanalytical Methods, Scand. J. Metall., 1979, 8(2), p 51–54
R. Pompe, A Thermodynamic Analysis of the Solubility of Nitrogen Gas in Manganese in the Range 13-25 at.%N, J. Less Common Metals, 1979, 65(2), p 237–252
A.D. Pelton, S.A. Degterov, G. Eriksson, C. Robelin, and Y. Dessureault, The Modified Quasichemical Model I-Binary Solutions, Metall. Mater. Trans. B, 2000, 31(4), p 651–659
A.D. Pelton and P. Chartrand, The Modified Quasi-chemical Model: Part II. Multicomponent Solutions, Metall. Mater. Trans. A, 2001, 32(6), p 1355–1360
M. Hillert, The Compound Energy Formalism, J. Alloys Compd., 2001, 320(2), p 161–176
M. K. Paek, Ph.D. Thesis, Hanyang University, 2014
M.K. Paek, S. Chatterjee, J.J. Pak, and I.H. Jung, Thermodynamics of Nitrogen in Fe-Mn-Al-Si-C Alloy Melts, Metall. Mater. Trans. B, 2016, 47(2), p 1243–1262
C.W. Bale, P. Chartrand, S.A. Degterov, G. Eriksson, K. Hack, R.B. Mahfoud, and S. Petersen, FactSage Thermochemical Software and Databases, Calphad, 2002, 26(2), p 189–228
A.T. Dinsdale, SGTE Data for Pure Elements, Calphad, 1991, 15(4), p 317–425
J. Leitner, P. Voňka, D. Sedmidubsky, and P. Svoboda, Application of Neumann–Kopp Rule for the Estimation of Heat Capacity of Mixed Oxides, Thermochim. Acta, 2010, 497(1–2), p 7–13
G. Inden, Project Meeting CALPHAD V, Max-Planck-Inst, Eisenforschung, GmbH, Dusseldorf, 1976, p 111
M. Hillert and M. Jarl, A Model for Alloying in Ferromagnetic Metals, Calphad, 1978, 2(3), p 227–238
A.D. Pelton and A. General, Geometric, Thermodynamic Model for Multicomponent Solutions, Calphad, 2001, 25(2), p 319–328
C. Wagner, Thermodynamics of Alloys, Addision-Wesley Press, Cambridge, 1952, p 47–51
K. Frisk, A New Assessment of the Fe-N Phase Diagram, Calphad, 1987, 11(2), p 127–134
D.R. Gaskell, Introduction to the Thermodynamics of Materials, 4th ed., Taylor & Francis, New York, 2003, p 131–132
M. Hillert and M.A. Jarl, Thermodynamic Analysis of the Iron-Nitrogen System, Metall. Trans. A, 1975, 6(3), p 553
J. Ågren, A Thermodynamic Analysis of the Fe-C and Fe-N Phase Diagrams, Metall. Trans. A, 1979, 10(12), p 1847–1852
J. Kunze, Thermodynamic Calculation of Phase Diagrams in the Iron-Nitrogen System, Steel Res. Int., 1986, 57(8), p 361–367
A. Guillermet Fernandez and v Du, Thermodynamic Analysis of the Fe-N System Using the Compound-Energy Model with Predictions of the Vibrational Entropy, Z. Metallkd., 1994, 85(3), p 154–163
H.A. Wriedt, N.A. Gokcen, and R.H. Nafziger, The Fe-N (Iron-Nitrogen) System, B. Alloy Phase Diagr., 1987, 8(4), p 355–377
A. Sieverts and G. Zapf, Iron and Nitrogen, Z. Phys. Chem., 1935, 172A, p 314–315
V.C. Kashyap and N. Parlee, Solubility of Nitrogen in Liquid Iron and Iron Alloys, Trans. Metall. Soc. AIME, 1958, 212(1), p 86–91
R.D. Pehlke and J.F. Elliott, Solubility of Nitrogen in Liquid Iron Alloys. I. Thermodynamics, Trans. Am. Inst. Min. Metall. Pet. Eng., 1960, 218(6), p 1088–1101
F. Ishii, S. Banya, and T. Fuwa, Solubility of Nitrogen in Liquid Iron and Iron Alloys Containing the Group VIa Elements, Tetsu-Hagane, 1982, 68(8), p 946–955
J. Chipman and D.W. Murphy, Solubility of Nitrogen in Liquid Iron, Tech. Publ. Am. Inst. Min. Metall. Eng., 1935, 591, p 12
W.Y. Kim, J.G. Kang, C.H. Park, J.B. Lee, and J.J. Pak, Thermodynamics of Aluminum, Nitrogen and AlN Formation in Liquid Iron, ISIJ Int., 2007, 47(7), p 945–954
D.H. Kim, M.S. Jung, H. Nam, M.K. Paek, and J.J. Pak, Thermodynamic Relation between Silicon and Aluminum in Liquid Iron, Metall. Mater. Trans. B, 2012, 43(5), p 1106–1112
D.W. Gomersall, A. McLean, and R.G. Ward, Solubility of Nitrogen in Liquid Iron and Liquid Iron-carbon Alloys, Trans. Metall. Soc. AIME, 1968, 242(7), p 1309–1315
R.P. Yadav and S.P. Pednekar, Solubility of Nitrogen in Liquid Iron and Iron-Carbon Alloys Using Levitation Melting Technique, Metall. Eng. IIT Bombay, 1971, 2, p 14–17
Y. Tsu and T. Saito, Absorption of Nitrogen by Molten Iron and Manganese at High Temperatures, Tohoku Daigaku Senko Seiren Kenkyusho Iho, 1971, 27(1–2), p 151–160
S.W. Lee and H.Y. Young, Effect of Manganese on the Nitrogen Solubility in an Iron Melt, Taehan Kumsok Hakhoechi, 1984, 22(9), p 789–793
M.K. Paek, J.M. Jang, K.H. Do, and J.J. Pak, Nitrogen Solubility in High Manganese-aluminum Alloyed Liquid Steels, Met. Mater. Int., 2013, 19(5), p 1077–1081
K.G. Leewis and A. McLean, Thermodynamics of Nitrogen Dissolution in Liquid Iron-Silicon Alloys, Can. Metall. Q., 1979, 18(3), p 333–340
H. Schenck, M.G. Frohberg, and H. Heinemann, Investigations on Nitrogen Absorption in Liquid Iron Alloys to a Pressure of Four Atmospheres, Arch. Eisen, 1962, 33, p 593–600
Y. Kasamatsu and S. Matoba, Activity of Nitrogen in Liquid Pure Iron, Tetsu-to-Hagane, 1959, 45(2), p 100–105
S. Maekawa and Y. Nakagawa, Solubility of Nitrogen in Liquid Iron and Effect of Carbon, Silicon and Manganese on the Solubility, Tetsu-to-Hagané, 1960, 46(7), p 748–753
A.H. Satir-Kolorz and H.K. Feichtinger, On the Solubility of Nitrogen in Liquid Iron and Steel Alloys Using Elevated Pressure, Z. Metallkd., 1991, 82(9), p 689–697
H. Feichtinger, A. Satir-Kolorz, and X.H. Zheng, Solubility of Nitrogen in Solid and Liquid Iron Alloys with Special Regard to the Melting Range, High Nitrogen Steels HNS, 1988, 88, p 75–80
W.Y. Kim, C.O. Lee, C.W. Yun, and J.J. Pak, Effect of Chromium on Nitrogen Solubility in Liquid Fe-Cr Alloys Containing 30 Mass% Cr, ISIJ Int., 2009, 49(11), p 1668–1672
T.I. Chung, J.B. Lee, J.G. Kang, J.O. Jo, B.H. Kim, and J.J. Pak, Thermodynamic Interactions of Nb and Mo on Ti in Liquid Iron, Mater. Trans., 2008, 49(4), p 854–859
H. Schenck, M.G. Frohberg, and H. Graf, Influencing the Equilibrium of Nitrogen with Liquid Iron Solutions by the Addition of Other Elements. II, Arch. Eisen, 1959, 30, p 533–537
L.S. Darken, R.P. Smith, and E.W. Filer, Solubility of Gaseous Nitrogen in Gamma Iron and the Effect of Alloying Constituents-Aluminum Nitride Precipitation, JOM, 1951, 3(12), p 1174–1179
N.S. Corney and E.T. Turkdogan, The Effect of Alloying Elements on the Solubility of Nitrogen in Iron, J. Iron Steel Inst., 1955, 180, p 334–348
J.D. Fast and M.B. Varrijp, Solubility of Nitrogen in Alpha-iron, J. Iron Steel Inst., 1955, 180, p 337–343
R.W. Fountain and J. Chipman, Solubility and Precipitation of Vanadium Nitride in Alpha and Gamma iron, Trans. TMS-AIME, 1958, 212, p 737–748
H.A. Wriedt and O.D. Gonzalez, The Solubility of Nitrogen in Solid Iron-nickel Alloys near 1000 °C, Trans. TMS-AIME, 1961, 221(3), p 532–535
T. Mori, K. Shinmyo, E. Ichise, and S. Koyama, Effects of Alloying Elements on the Activity of Nitrogen in Austenite. II, Effect of Nickel and Solubility of Nitrogen in Pure Iron, Mem. Fac. Eng. Kyoto Univ., 1963, 25, p 175–187
H. Schenck, M.G. Frohberg, and F. Reinders, The Solubility of Nitrogen in Iron Alloys in the Temperature Range 700 °C to 1200 °C, Stahl Eisen, 1963, 83(2), p 93–99
P. Grieveson and E.T. Turkdogan, Kinetics of Reaction of Gaseous Nitrogen with Iron Part II: Kinetics of Nitrogen Solution in Alpha and Delta Iron, Trans. TMS-AIME, 1964, 230, p 407–414
I.N. Milinskaya and J.A. Tomlin, Solubility and Heat of Solution of Nitrogen in Iron, Akad. Nauk SSSR Doklad., 1967, 174(1), p 135–138
D. Atkinson and C. Bodsworth, Thermodynamic Properties of Nitrogen in Austenitic Iron and Iron-Nickel Alloys, J. Iron Steel Inst., 1970, 208(6), p 587–593
H. Zitter and L. Habel, Solubility of Nitrogen in Pure Iron and in Austenitic Chromium-Nickel-Steels, Arch. Eisen, 1973, 44(3), p 181–188
R.B. McLellan and R.J. Farraro, Thermodynamics of the Iron-Nitrogen System, Acta Metall., 1980, 28(3), p 417–422
J. Kunze and K. Friedrich, Equilibrium between Gaseous Nitrogen and Nitrogen Dissolved in Iron, Neue Hutte, 1984, 29(10), p 378–382
M.A.J. Somers, B.J. Kooi, L. Maldzinski, E.J. Mittemeijer, A.A. Van Der Horst, A.M. Van Der Kraan, and N.M. Van Der Pers, Thermodynamics and Long-Range Order of Interstitials in an HCP Lattice: Nitrogen in ε-Fe2N1-z, Acta Mater., 1997, 45(5), p 2013–2025
H. Schenck, M.G. Frohberg, and W. Kranz, Investigations on the Solubility of Nitrogen in Iron-Manganese Alloys at 1000 C and Pressures up to 65 atm, Arch Eisen., 1963, 34(11), p 825–830
M. Naka, T. Masumoto, and Y. Imai, Solubility of Nitrogen in Austenitic Iron under High Nitrogen Pressure and Thermodynamic Properties of Iron-Nitrogen Interstitial Solid Solution, Sci. Rep. Res. Inst. Tohoku Univ. Ser. A Phys. Chem Metall., 1972, 24, p 30–47
A.P. Bashchenko, V.I. Isotov, A.V. Omelchenko, V.I. Soshnikov, and G.V. Shcherbedinski, Alloying Iron and Its Alloys with Nitrogen Under High Pressure, Russ. Metall., 1985, 4, p 169–174
E. Lehrer, Magnetic Investigation of the System Iron-Nitrogen, Z. Elektrochem., 1930, 36(7), p 460–473
L.J. Dijkstra, Precipitation Phenomena in the Solid Solutions of Nitrogen and Carbon in Alpha Iron Below the Eutectoid Temperature, JOM, 1949, 1(3), p 252–260
V.G. Paranjpe, M. Cohen, M.B. Bever, and C.F. Floe, The Iron-Nitrogen System, JOM, 1950, 2(2), p 261–267
A. Burdese, Research on Fe-N System, Metall. Ital., 1955, 47, p 357–361, 366
R. Rawlings and D. Tambini, The Determination of the Alpha Phase Boundaries of the Iron-Nitrogen System by Internal Friction Methods, J. Iron Steel Inst., 1956, 184(11), p 302–308
W. Pitsch and E. Houdremont, A Contribution to Fe-N System, Arch. Eisen, 1956, 27, p 281–284
M. Nacken and J. Rahmann, Quench Aging of Nitrogen-containing Steel by Measuring Damping, Coercive Field Strength, and Recovery, Arch. Eisen, 1962, 33(2), p 131–140
H. A. Wriedt, L. S. Darken, and R. P. Smith, The Thermodynamics of Nitrogen in α-Fe at 300 to 600 °C, 1964, Unpublished
Y. Imai, T. Masumoto, and M. Sakamoto, Effect of Nickel on the Solubility and Diffusion of Nitrogen in α-Fe, Sci. Rept. Res. Inst. Tohoku Univ. Ser. A, 1968, 20(1), p 1–13
K. Abiko and Y. Imai, The Precipitation of Nitrogen during Isothermal Aging in a Quenched Fe-N Alloy, Trans. Jpn. Inst. Metals, 1977, 18(2), p 113–124
Z. Przylecki, NH3-H2-Fe-N System Equilibrium Part I: The Stability of Ferrite and Austenite, Sesja Naukowa Kom. Techn. Budowy Maszyn, 1979 (Poznan), p 35–84.
O. Eisenhut and E. Kaupp, Radiographic Investigation of the System, Iron-Nitrogen, Z. Elektrochem. Angew. Phys. Chem., 1930, 36, p 392–404
S. Brunauer, M.E. Jefferson, P.H. Emmett, and S.B. Hendricks, Equilibria in the Iron-Nitrogen System, J. Am. Chem. Soc., 1931, 53(5), p 1778–1786
K.H. Jack, The Iron-Nitrogen System: The Preparation and the Crystal Structures of Nitrogen-Austenite (γ) and Nitrogen-Martensite(α’), Proc. R. Soc. A, 1093, 1951(208), p 200–215
E. Scheil, W. Mayr, and J. Mueller, Production of Iron-Carbon-Nitrogen Alloys and Determination of Equilibriums in this System, Arch. Eisen, 1962, 33(6), p 385–392
J. Stein, R.E. Schacherl, M. Jung, S. Meka, B. Rheingans, and E.J. Mittemeijer, Solubility of Nitrogen in Ferrite; the Fe-N Phase Diagram, Int. J. Mater. Res., 2013, 104(11), p 1053–1065
T. Woehrle, H. Cinaroglu, A. Leineweber, and E.J. Mittemeijer, Fe-N and Fe-N-C Phase Equilibria above 853 K Studied by Nitriding/Nitrocarburising and Secondary Annealing, Int. J. Mater. Res., 2016, 107(3), p 192–202
H.J. Grabke, Defects in γ-iron Nitride, Berichte der Bunsen-Gesellschaft, 1969, 73(6), p 596–601
H.A. Wriedt, Thermodynamics of Gamma Prime Iron Nitride at 500 °C, Trans. Metall. Soc. AIME, 1969, 245(1), p 43–46
Z. Przylecki and L. Maldzinski, Conditions of Equilibrium Between Gas Mixture NH3-H2 and Nitrogen in Nitride γ, Azotki, 1987, 1, p 153–162
E.D.M. Van Voorthuysen, D.O. Boerma, and N.C. Chechenin, Low-Temperature Extension of the Lehrer Diagram and the Iron-Nitrogen Phase Diagram, Metall. Mater. Trans. A, 2002, 33(8), p 2593–2598
L. Małdzinski, Z. Przyłȩcki, and J. Kunze, Equilibrium Between Ammonia-Hydrogen Mixtures and the ε Phase of Iron, Steel Res. Int., 1986, 57(12), p 645–649
P.H. Emmett, S.B. Hendricks, and S. Brunauer, The Dissociation Pressure of Fe4N, J. Am. Chem. Soc., 1930, 52(4), p 1456–1464
A. Burdese, Research on the Heat of Formation and on the Magnetic Properties of Iron Nitrides, Ann. Chim., 1959, 49, p 1873–1884
H.H. Podgurski and F.N. Davis, Thermochemistry and Nature of Nitrogen Absorption in Nitrogenated Fe-Ti Alloys, Acta Metall., 1981, 29(1), p 1–9
I.B. Baratashvili, V.P. Fedotov, A.M. Samarin, and V.M. Berezhiani, The Solubility of Nitrogen in Liquid Manganese, Dokl. Akad. Nauk. SSSR, 1961, 139(6), p 1354–1355
S.Z. Beer, Solubility of Nitrogen in Molten Iron-manganese Alloys, Trans. Am. Inst. Min. Metall. Pet. Eng., 1961, 221(1), p 2–8
N.A. Gokcen, Solubility of Nitrogen in Liquid Manganese, Trans. Metall. Soc. AIME, 1961, 221(1), p 200–201
V.P. Perepelkin, The Solubility of Nitrogen in Melts of Standard Metallic Manganese, Izv. Vysshikh Uchebnykh Zaved. Chernaya Metall., 1966, 9(3), p 88–93
G.M. Grigorenko, Y.M. Pomarin, V.I. Lakomskii, and A.V. Sherevera, Effect of Temperature on the Solubility of Nitrogen in Iron-manganese Melts, Izv. Akad. Nauk. SSSR Met., 1974, 6, p 11–15
G.J.W. Kor, Solubility of Nitrogen in Liquid Manganese, Metall. Trans. B-Pro. Metall., 1978, 9(1), p 97–99
E.J. Kim, J.J. Pak, and B.D. You, Nitrogen Solubility in Liquid Manganese and Ferromanganese Alloys, Metall. Mater. Trans. B, 2001, 32(4), p 659–668
J.M. Jang, J.Y. Eom, M. Jiang, M.K. Paek, and J.J. Pak, Nitrogen Solubility in Liquid Manganese Alloys Containing Silicon, Iron and Carbon, ISIJ Int., 2013, 53(5), p 768–773
P. Ettmayer, A. Vendl, E. Horvath, and R. Kieffer, Contribution to the Study of the CHROMIUM-MANGANESE-NITROGEN System, Monatsh. Chem., 1978, 109(6), p 1277–1285
O. Prelinger, Nitrogen Compounds of Manganese, Monatsh. Chem., 1894, 15, p 391–401
U. Zwicker, The System Mn-N, Z. Metallkd., 1951, 42, p 277–278
GSh Mamporiya and R.I. Agladze, Nitridation of Manganese by Ammonia, Zh. Neorg. Khim., 1967, 12(10), p 2541–2545
H. Jacobs and C. Stueve, High-Pressure Synthesis of the η-Phase in the Manganese-nitrogen System: Manganese Nitride(Mn3N2), J. Less Common Metals, 1984, 96, p 323–329
G. Kreiner and H. Jacobs, Magnetic Structure of η-Manganese Nitride(η-Mn3N2), J. Alloys Compd., 1992, 183, p 345–362
M. Tabuchi, M. Takahashi, and F. Kanamaru, Relation Between the Magnetic Transition Temperature and Magnetic Moment for Manganese Nitrides MnNy (0 < y<1), J. Alloys Compd., 1994, 210(1–2), p 143–148
A. Leineweber, R. Niewa, H. Jacobs, and W. Kockelmann, The Manganese Nitrides η-Mn3N2 and θ-Mn6N5+x: Nuclear and Magnetic Structures, J. Mater. Chem., 2000, 10(12), p 2827–2834
K. Suzuki, T. Kaneko, H. Yoshida, Y. Obi, H. Fujimori, and H. Morita, Crystal Structure and Magnetic Properties of the Compound MnN, J. Alloys Compd., 2000, 306(1–2), p 66–71
F. Tessier, M.R. Ranade, A. Navrotsky, R. Niewa, F.J. DiSalvo, A. Leineweber, and H. Jacobs, Thermodynamics of Formation of Binary and Ternary Nitrides in the System Ce/Mn/N, Z. Anorg. Allg. Chem., 2001, 627(2), p 194–200
H. Yang, H. Al-Brithen, E. Trifan, D.C. Ingram, and A.R. Smith, Crystalline Phase and Orientation Control of Manganese Nitride Grown on MgO(0 0 1) by Molecular Beam Epitaxy, J. Appl. Phys., 2002, 91(3), p 1053–1059
W.R. Lambrecht, M. Prikhodko, and M.S. Miao, Electronic Structure and Magnetic Interactions in MnN and Mn3N2, Phys. Rev. B, 2003, 68(17), p 174411
H. Yang, R. Yang, A.R. Smith, and W.R. Lambrecht, Atomic-scale Structure of η-Phase Mn3N2(0 1 0) Studied by Scanning Tunneling Microscopy and First-Principles Theory, Surf. Sci., 2004, 548(1–3), p 117–128
A. Kędziorski and M.C. Munoz, Magnetic Configuration, Electronic Structure, and Stability of the Low-Index Surfaces of η-Mn3N2: A First-Principles Study, Phys. Rev. B, 2012, 86(15), p 155–455
R. Yu, X. Chong, Y. Jiang, R. Zhou, W. Yuan, and J. Feng, The Stability, Electronic Structure, Elastic and Metallic Properties of Manganese Nitrides, RSC Adv., 2015, 5(2), p 1620–1627
R. Schenck and A. Kortengraber, The System: Manganese-nitrogen, Z. Anorgan. Allg. Chem., 1933, 210, p 273–285
N. Ōtsuka, Y. Hanawa, and S. Nagakura, Crystal Structure and Phase Transition of Mn6N5 Studied by Electron Diffraction, Phys. Status Solidi(a), 1977, 43(2), p K127–K129
L. Kaufman, Coupled Phase Diagrams and Thermochemical Data for Transition Metal Binary Systems-III, Calphad, 1978, 2(2), p 117–146
H. Weiming, An Assessment of the Fe-Mn System, Calphad, 1987, 11(2), p 183–186
R. Kubitz and F.H. Hayes, Enthalpies of Mixing in the Iron-manganese System by Direct Reaction Calorimetry, Monatsh. Chem./Chem. Monthly, 1987, 118(1), p 31–41
V.T. Witusiewicz, F. Sommer, and E.J. Mittemeijer, Enthalpy of Formation and Heat Capacity of Fe-Mn Alloys, Metall. Mater. Trans. B, 2003, 34(2), p 209–223
V.T. Witusiewicz, F. Sommer, and E.J. Mittemeijer, Reevaluation of the Fe-Mn Phase Diagram, J. Phase Equilib. Diffusion, 2004, 25(4), p 346–354
J. Kunze, P. Brož, J. Sopoušek, and W. Gruner, Thermodynamic Investigation of the Austenite and the Delta Ferrite in the System Fe-Cr-Mn-N, Steel Res., 1996, 67(1), p 26–33
J. Kunze, P. Broz, and I. Stloukal, Thermodynamic Analysis of the δ Ferrite in the Systems Fe-Cr-N and Fe-Cr-Mn-N, Steel Res., 1996, 67(7), p 279–284
W. Wentrup and O. Rief, The Solubility of Nitrogen in Iron Melts with Chromium, Manganese, and Nickel Additions, Arch. Eisen, 1949, 20(11–12), p 359–362
F. Ishii, S. Ban-ya, and T. Fuwa, Solubility of Nitrogen in Liquid Iron Alloys, Tetsu-to-Hagané, 1982, 68(10), p 1551–1559
H. Wada, S.W. Lee, and R.D. Pehlke, Nitrogen Solubility in Liquid Fe and Fe-Mn Alloys, Metall. Mater. Trans. B, 1986, 17(1), p 238–239
J.H. Shin, J. Lee, D.J. Min, and J.H. Park, Solubility of Nitrogen in High Manganese Steel (HMnS) Melts: Interaction Parameter Between Mn and N, Metall. Mater. Trans. B, 2011, 42(6), p 1081–1085
R.A. Dodd and N.A. Gokcen, Solubility of Nitrogen in Liquid-Iron Manganese Alloys, Trans. Metall. Soc. AIME, 1961, 221(2), p 233–236
J.M. Jang, M.K. Paek, and J.J. Pak: Hanyang University, Ansan, South Korea, 2016, Unpublished Research.
J. Rawers and N. Gokcen, Nitrogen Concentration in Liquid Fe-Mn Alloys at High Pressure, J. Phase Equilib., 1993, 14(2), p 179–183
H.J. Grabke, S.K. Iyer, and S.R. Srinivasan, Solubility of Nitrogen in Austenitic Iron-Manganese and Iron-Chromium Alloys, Z. Metallkd., 1975, 66(5), p 286–292
K. Iwase and M. Fukusima, Absorption of Hydrogen by Some Metals and Their Alloys with Iron, Nippon Kinzoku Gakkaishi, 1937, 1, p 202–213
J. Kunze and I. Rothe, Solubility of Nitrogen in Austenitic FeCrMn Alloys, Steel Res. Int., 1994, 65(8), p 331–337
C. Ko and R.B. Molellan, Thermodynamic of Ternary Nitrogen Austenites, Acta Metall., 1983, 31, p 1821–1827
J.F. Enrietto, Solubility and Precipitation of Nitrides in Alpha-Iron Containing Manganese, Trans. Metall. Soc. AIME, 1962, 224(1), p 43–48
M. Nacken and U. Kuhlmann, Effect of Manganese Content on the Precipitation Behavior of Nitrogen-Containing Ferrous Alloys, Arch. Eisen, 1966, 37(4), p 331–339
R. Rawlings and P.G. Hatherley, Iron-Manganese-Nitrogen Ferrite: The Activity of Nitrogen and the Solubility of Manganese Nitrides, Metal Science, 1975, 9(1), p 97–103
A. Burdese, D. Firrao, P. Rolando, and M. Rosso, Iron-Manganese-Nitrogen System, La Chim. E L’Ind., 1984, 66(7–8), p 456–460
Acknowledgment
We would like to gratefully acknowledge the Steelmaking Consortium Members, Posco, Hyundai Steel, Doosan Heavy, Industry and Construction, Nucor Steel, RioTinto Iron and Titanium, Nippon Steel and Sumitomo Metals Corp., JFE Steel, Voestalpine, RHI, Tata Steel Europe, and Natural Science and Engineering Research Council of Canada for supporting the present project. This work was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF- 2015R1A5A1037627).
Author information
Authors and Affiliations
Corresponding author
Additional information
This invited article is part of a special issue of the Journal of Phase Equilibria and Diffusion in honor of Prof. Zhanpeng Jin’s 80th birthday. The special issue was organized by Prof. Ji-Cheng (JC) Zhao, The Ohio State University; Dr. Qing Chen, Thermo-Calc Software AB; and Prof. Yong Du, Central South University.
Rights and permissions
About this article

Cite this article
You, Z., Paek, MK. & Jung, IH. Critical Evaluation and Optimization of the Fe-N, Mn-N and Fe-Mn-N Systems. J. Phase Equilib. Diffus. 39, 650–677 (2018). https://doi.org/10.1007/s11669-018-0666-8
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11669-018-0666-8