
Abstract
High-temperature applications have demonstrated aluminide-coated nickel-base superalloys to be remarkably effective, but are reaching their service limit. Alternate materials such as refractory (e.g., W, Mo) silicide alloys and SiC composites are being considered to extend high temperature capability, but the silica surfaces on these materials require coatings for enhanced environmental resistance. This can be accomplished with a Mo-Si-B-based coating that is deposited by a spray deposition of Mo followed by a chemical vapor deposition of Si and B by pack cementation to develop an aluminoborosilica surface. Oxidation of the as-deposited (Si + B)-pack coatings proceeds with partial consumption of the initial MoSi2 forming amorphous silica. This Si depletion leads to formation of a B-saturated Mo5Si3 (T1) phase. Reactions between the Mo and the B rich phases develop an underlying Mo5SiB2 (T2) layer. The T1 phase saturated with B has robust oxidation resistance, and the Si depletion is prevented by the underlying diffusion barrier (T2). Further, due to the natural phase transformation characteristics of the Mo-Si-B system, cracks or scratches to the outer silica and T1 layers can be repaired from the Si and B reservoirs of T2 + MoB layer to yield a self-healing characteristic. Mo-Si-B-based coatings demonstrate robust performance up to at least 1700 °C not only to the rigors of elevated temperature oxidation, but also to CMAS attack, hot corrosion attack, water vapor and thermal cycling.
Similar content being viewed by others


Introduction
The continuing drive to attain increased gas turbine alloys and SiC composites engine performance in terms of output power along with reduced emissions and improved efficiency requires the incorporation of new high-temperature materials with capabilities beyond the limitations of current Ni-base superalloys (Ref 1). Both multiphase Mo- and Nb-base refractory metal alloys and SiC composites are receiving increased attention since they offer high temperature strength and creep resistance (Ref 2). While the Mo-Si-B alloys have some intrinsic oxidation resistance, the level is insufficient for operational conditions (Ref 3). In order to provide the supplemental environmental resistance, there is a clear trend to rely on coatings that must exhibit a robust and versatile performance so that they can endure a range of aggressive environments without degradation (Ref 4, 5). For example, in a high temperature combustion environment about 10% is water vapor that attacks pure chromia and silica to form volatile products (Ref 6). Similarly, salt ingestion and fuel impurities such as S and V result in hot corrosion and fluxing of surface protective layers and CMAS (calcia, magnesia aluminosilica) attacks thermal barrier coatings (Ref 7). With this variety of aggressive environments, it is a challenge to identify one coating that will provide the required protection.
The use of thermal spray processing has shown utility in a wide range of applications: coatings for wear, thermal or corrosion resistance as well as environmental barriers. These take the form of either single coating layers or as graded multilayers (Ref 8). The breadth of uses for these methods reflects the processing flexibility of the technology. In addition to the use of thermal spray technologies for direct coating synthesis, they can also provide a valuable capability in developing a precursor coating that is converted subsequently into a final robust coating structure (Ref 9). To demonstrate the method of thermal spraying a precursor material, an example two-step synthesis of the Mo-Si-B coating on a refractory ceramic composite is presented along with the coating performance in several aggressive environments.
Coating and Substrate Preparation
Recent developments in environmental resistant coatings involve the implementation of a Mo-Si-B-based system for protection at a range of elevated temperatures (800-1500 °C) in ambient air. In the initial development on Mo-base alloys, a co-deposition of Si and B via pack cementation yielded an aluminoborosilica outer layer. Subsequent treatment by exposure in air at elevated temperature (1300-1450 °C) resulted in the final coating structure. For other substrates without Mo such as cermets, SiC composites and ZrB2-SiC ceramics, the coating is applied with a two-step method: (1) chemical vapor deposition (CVD) of Mo by the decomposition of Mo(CO)6 at 225 °C; and (2) pack cementation, a CVD process, of Si and B (35 wt.% Si:1 wt.% B ratio) under Ar at 1000 °C. Depositing Mo through the decomposition of Mo(CO)6 results in complete surface coverage of the substrate, but the final Mo layer is less than 10 µm thick. This limits the total thickness of the oxidation-resistant coating and thus the Si and B reservoir size and, ultimately, the level of protection. The slow deposition rate and production of CO gas (Ref 10) constrain the process to laboratory scale only. Additionally, many passes of the Mo CVD process are required to coat substrates with complex geometries, showing that this method has little opportunity to be scaled up to industrial standards. A possible alternative is to develop a plasma spray method to apply a surface molybdenum layer on an arbitrary substrate (Ref 9). Compared with the Mo deposition by the CVD carbonyl method, plasma spraying of Mo enables thicker coatings on larger and more geometrically complicated samples. This enables the Mo-Si-B coating be applied to a wider range of end uses and to be scaled up beyond laboratory scales. Plasma spraying deposition works by creating an arc jet and accelerating it through a nozzle. This arc is used to heat the material to be deposited on the substrate. This plasma spray method can achieve sufficiently high deposition rates of refractory metals to be industrially relevant while leaving the substrate undamaged (Ref 8).
Spraying Operations
For substrates without Molybdenum, a layer of Mo was applied via plasma spraying. The surfaces of the samples were first prepared using a grit blast of 120 grit alumina. Next the samples were preheated to 204 °C in preparation of the thermal spray process. The source of the Mo to be deposited was as-received H. C. Starck Amperit 105 powder: 99.5% agglomerated and sintered, a particle size range of 18-57 µm and a measured density of 2.25 g/cm3. A Thermach SG-100 gun was used in the single-pass plasma spraying method, and a Fanuc M-16ib robot was employed for controlled and repeatable application. An Ar/He gas combination was used in the Arc Plasma Spraying process with an argon carrier gas. Table 1 contains additional spraying parameters. The parameters listed in Table 1 represent the optimum conditions for a high coating deposition efficiency, including a low oxide content.
Coating Synthesis and Structure
Following the plasma spray deposition of a precursor layer of Mo on to Mo-based substrates, subsequent co-depositions of Si and B were applied by pack cementation. The coating process is demonstrated schematically in Fig. 1. The pack powder is comprised of both Si and B as the deposition source that is effective within a mixture of 35 wt.% Si:1 wt.% B to 20 wt.% Si:1 wt.% B, Al2O3 powder filler and a halide activator such as NaF or NH4Cl.

Schematics of the two-stage coating synthesis method. Stage I is plasma spraying of Mo onto the substrate; Stage II is the CVD process of Si and B co-deposition (reproduced from Ref 9)
The plasma-sprayed samples were then surrounded by the powder on all sides inside of an alumina boat. The boat was placed in an alumina tube furnace that was then sealed and evacuated, in order to remove oxygen from the system. Pack cementation was performed at 1000 °C under an atmosphere of argon flowing through the system for times ranging from 90 min to 50 h. The details of the pack cementation method are outlined elsewhere (Ref 11-13).
Following the pack cementation, a process of conditioning annealing treatments at 1200-1400 °C for times from 10 to 30 h (Ref 4, 14) is required to reach the mature coating structure. Indeed, as shown in Fig. 2(a), the B content in the pack powder mixture stabilizes a continuous layer of MoB underneath the silicide layer, between the Mo alloy substrate and the rest of the coating. As illustrated in Fig. 2(b), the nominal composition trajectory for the co-deposition processes can be illustrated by rotation about the nominal substrate composition whereby the vapor source begins as pure Si before moving to a mixed Si/B source.

(a) As-packed microstructure of Mo-Si-B coating. (b) Composition trajectory of Si + B pack cementation process on Mo-3Si-1B (wt.%) alloys depicted in the Mo-Si-B phase diagram. A full development of the boride phase underneath the silicide phase is observed. (c) Typical post-conditioning microstructure of the Mo-Si-B coating. An amorphous oxide forms on top of a MoSi2 and Mo5Si3 two phase region, which in turn covers a layer of Mo5Si3 and Mo5SiB2, which acts as a diffusion barrier between the coating and the substrate (Fig. 2(b) is adapted from Ref 4)
During oxidation exposures of the (B + Si)-pack alloys, the initial MoSi2 is transformed into the T1 phase as a result of the transient composition trajectory in Fig. 2(b). With continued high-temperature oxidation, the outer T1 phase layer eventually will be consumed by dissolution into the substrate. However, the inward flux of Si and B that results in the formation of T1 also leads to the growth of the T2 borosilicide and/or boride phase layer. The B/Si ratio of the pack cementation powder source determines the relative amounts of the T2 and MoB phases below the T1 phase region. This is illustrated in the composition trajectories presented in Fig. 2(b). Due to the phase equilibria, the T1 phase is always in contact with the MoB or T2 borosilicide phase, which ensures B saturation in the T1 phase. Furthermore, due to the excellent oxidation resistance of B-saturated T1 (Ref 15, 16) the loss of Si is blocked by the underlying diffusion barrier (i.e., the borosilicide and boride phases). This has the net effect of arresting thickness change of the T1 layer (Ref 4, 14). Further, any damage to the outer T1 layer can be repaired from the T2 + MoB layer below. In effect, the same reaction that yields the T2 + MoB layer additionally provides a kinetic bias (Ref 5) that allows for the continued existence of the outer T1 layer and also results in a self-healing characteristic of the coating.
Oxidation tests performed at 1400 °C show the effective oxidation protection yielded by the coatings. As shown in Fig. 3(a), with uncoated samples, oxidation of Mo-3Si-1B (wt.%) alloys causes a large mass loss as shown by the thickness reduction in the samples. The cross-sectional SEM image of the oxide scales shows that although there is a thick and continuous borosilicate layer on the surface, the high evaporation rate of B2O3 as well as MoO3 at this temperature results in a relatively high recession rate during the transient oxidation. In contrast, the coated sample shows the thickness retention of the sample and furthermore sluggish growth of the borosilicate layer on top of the coating (less than 15 μm after 30-h exposure).

(a) Oxidation of an uncoated sample of Mo-3Si-1B (wt.%) in air at 1400 °C for 10 h results in a large mass loss as evident by the thickness reduction in the sample. The SEM cross section of the oxide scales shows that even though there is a thick and continuous borosilica layer on the surface, the high evaporation rate of B2O3 at this temperature results in a relatively large mass loss during the transient oxidation. (b) In contrast, the coated sample subjected to the same exposure in air at 1400 °C for 10 h shows thickness retention and very limited growth of the borosilica layer on top of the coating (adapted from Ref 4)
Water Vapor Exposure
Water vapor is known to accelerate oxidation attack in a combustion environment. Silicon containing compounds are thusly attacked due to the formation of volatile silica hydroxides, which rapidly leave the system (Ref 6, 17-20). Previous work on the oxidation of silica by steam in a flowing gas stream has been successfully modeled as paralinear behavior that results in rapid recession of the surface (Ref 6, 17-20). With the addition of boron to the silica, additional hydroxide phases become relevant, some of which are stable, including BO(OH), [BO(OH)]3 and B(OH)3 (Ref 17, 21, 22). Boron exposed to wet oxidation has been observed to form volatile boron hydroxides in significant amounts at relatively modest temperatures (Ref 17). However, the effect of wet oxidation on borosilicide coatings, including Mo-Si-B-based coatings, has not been fully evaluated particularly at high temperature (>1400 °C).
Wet oxidation conditions cause uncoated Mo-Si-B alloys to demonstrate major mass losses. This is demonstrated in Fig. 4(a), where an uncoated Mo-Si-B alloy was subjected to wet air at 1400-1500 °C for 24 h. Steady-state oxidation is eventually reached, in which a borosilica surface layer develops at the rate observed in coated samples, but not before significant mass losses. The volatilization and loss of Mo via Mo oxides and/or Mo hydroxides coupled with a high relative B/Si ratio delayed the approach of steady-state oxidation (Ref 8). The coated samples instead have an initial borosilica surface layer as well as the underlying borosilicide phases. Figure 4(b) illustrates that these features protect against aggressive water vapor attack conditions. Moreover, a detailed study of the coating composition has elucidated that the glass contains residual Al and Na that was incorporated from the pack powder to form an aluminoborosilica glass (Ref 12).

SEM cross section of (a) uncoated (b) coated Mo-Si-B coupon exposed to wet oxidation showing the benefit of borosilicide coatings in minimizing the mass recession rate (adapted from Ref 5)
Coating pure Mo with Mo-Si-B probes solely the protective capabilities of the coating, thus removing any influence from intrinsic oxidation resistance of the alloy. The mass change per area plot in Fig. 5 illustrates the coating’s protective ability under conditions of water vapor attack at 1300 °C compared to an uncoated Mo-Si-B alloy in the same conditions at 1100 °C (Ref 23). Two sets of coated samples are shown, with conditioned samples being pre-oxidized in ambient air, as earlier outlined, prior to water vapor exposure. Unconditioned samples were instead exposed to water vapor immediately after pack cementation. The alloy exhibits initial mass losses during transient-stage oxidation before steady state; both the conditioned and unconditioned Mo-Si-B coatings instead show immediate mass gain and paralinear oxidation kinetics.

Experimental data and fitted model for conditioned and unconditioned sample oxidized at 1300 °C up to 100 h, showing significant improvements in performance vs. uncoated Mo-Si-B alloys and by conditioning
Both literature (Ref 23, 24) and the current work suggest that there are two primary factors that result in the minimal effect of water vapor on the high temperature oxidation performance of Mo-Si-B coatings. The water vapor effect on the Mo-Si-B alloys is significant during the transient stage of the oxidation process, causing increased recession rates and mass losses from the evaporating volatiles (both Mo hydroxides and MoO3). However, the current work demonstrates that the presence of a preexisting aluminoborosilica scale from the coating reduced the transient-stage oxidation considerably and thus water vapor attack is almost completely mitigated. In essence, the preexisting glass lowers the available oxygen partial pressure at the MoSiB coating/glass interface, sufficiently below the threshold to prevent the formation of either MoO3 or Mo hydroxide. Rather, solid-state transformation from Si-rich to more Mo-rich silicides for the production of SiO2 is favorable over concurrent MoO3(g) or MoO2(s) production, which occurs below an oxygen partial pressure of approximately 1.01 Pa (Ref 24). This can be ascribed to the continued presence of glass modifiers such as Al, Na and B within the silica. These can be detected even after exposure at temperature as high as 1650 °C (Ref 12, 25). The current study suggests that the additive retention benefits the coatings by lowering the effective mass loss rate of the silica-based glass.
Another major factor of water vapor attack described in previous studies is the process of accelerated crystallization of silica-based glass. The formation of cristobalite encourages porous structures that cause additional degradation. The current work shows that the glass produced from MoSiB coatings does not exhibit this particular water vapor behavior. However, there is an observable difference in the character of glass produced under water vapor conditions (unconditioned coatings) and of glass produced in ambient air prior to water vapor testing (conditioned coatings). The conditioned coating in Fig. 5 demonstrates a greater capability of resisting both oxidation and hydroxide formation when the initial protective oxide layer is established in ambient air, which is noted by the lower net mass gains and the extended timescale for the paralinear behavior of the conditioned sample. It is clear that the development of the continuous borosilica layer in the presence of water vapor alters its protective capabilities. Despite this observation, the glass formed during the water vapor exposures retains an amorphous structure. This is hypothesized to result from the additive retention during oxidation, but further study is required to verify this explanation.
Cyclic Oxidation Performance
For a complete evaluation of the layered coating structure performance, isothermal exposure is inadequate since the potential mechanical incompatibility due to different coefficients of thermal expansion of the layers can yield cracking and degradation during thermal cycling. The cyclic oxidation exposures were conducted at 1300 °C in air with a 10-h hold followed by furnace cooling between each cycle. Figure 6 depicts the normalized mass change results during the 500-h of oxidation exposure at 1300 °C. The two samples exposed inside an alumina crucible are represented with filled symbols, whereas the two samples that were placed on alumina-free SiC foam are shown with empty marks. All four samples exhibited material loss during the first 100 to 120 h of cycling. After 100 h of oxidation, one sample of each condition was cut and imaged in the SEM. The remaining two samples were further tested for an additional 400 h and experienced discontinuous mass gains. Even though the time duration was too short to demonstrate parabolic oxidation kinetics, the overall magnitude of mass change is quite slow for testing at 1300 °C; with a maximum value of +/−0.2 mg/cm2. An analysis of morphological changes and phase evolution as a result of the exposure can elucidate the cause for the mass loss and deviation from parabolic behavior. The microstructure, mass change and chemical composition exhibited difference between the sample holders, thus, do not have an effect on overall mechanism.

Cyclic mass change per surface area vs. time of the coated samples at 1300 °C. Note the small overall magnitude of mass change in the 500-h duration exposure (adapted from Ref 42)
Since the coating structure during Si+B pack cementation can be formed regardless of the base Mo alloy composition, the effectiveness of this method was applied to a Mo-rich alloy, TZM (typical TZM has three major alloying elements of 0.4-0.55 wt.% Ti, 0.06-0.12 wt.% Zr and 0.01-0.04 wt.% C). Both Si and B were deposited onto the TZM substrate by pack cementation. It is well known that there is a large coefficient of thermal expansion (CTE) mismatch between MoSi2 and the TZM alloys as illustrated in Fig. 7. Despite superb coating performance under isothermal conditions, this CTE mismatch is cause for concern when applying the coatings in a cyclic environment. In fact, it has been well documented that the disilicide (MoSi2) performs quite poorly when subjected to cyclic oxidation tests when is applied onto Mo. Significant mass losses are observed due to MoO3 volatilization. Large residual stress is generated during cooling that lead to cracks that propagate through the coating and cause accelerated degradation. However, as Fig. 7 indicates, in contrast to MoSi2, the CTE of the a axis for the T1 phase is a near match to that of TZM. In fact, it was found that MoSi2 within the coatings could be converted into a [001] textured T1 phase by a simply annealing (i.e., conditioning) the specimens as illustrated by the inverse pole figure on the growth direction as well as the pole figures orthogonal to several crystallographic planes (Fig. 8). The result of replacing the disilicide by the T1 phase on the cyclic oxidation resistance of the coatings has been demonstrated to yield an oxidation performance that approaches the excellent behavior observed in isothermal cases. This is partially owed to an absence of transverse cracking in the T1 phase due to good CTE matching. Moreover, the coated samples avoid pesting during oxidation at 700 °C (Ref 4, 5).

Coefficent of thermal expansion (CTE) (10−6/K) for TZM, MoSi2 (a and c) and T1 phase (c). The CTE values of TZM and T1 phase (a) are almost similar (reproduced from Ref 7)

(a) Inverse pole figure of the T1 phase coatings on the TZM alloys along the growth direction showing the [001] preference, (b)-(d) pole figures along several crystallographic planes confirming the texture (reproduced from Ref 7)
Arc Jet Exposure
The harsh conditions experienced by spacecraft during atmospheric reentry or aircraft during hypersonic flight result in extremes in temperature (>2000 °C) which require materials with ultra-high melting temperatures. Refractory metals or high melting temperature ceramics are the primary candidates for such challenging applications. Furthermore, because of aerodynamic heating the environment contains hot ionized gases, which cause more aggressive attack than oxidation in static air. In order to simulate these unique conditions, specialized facilities such as arc jet apparatuses are necessary. Most of the literature concerning arc jet testing focus on HfB2 and ZrB2 and their composites with SiC (Ref 26-29). The primary findings after relatively short exposure times on the order of minutes at temperatures up to 2000 °C are ablation and erosion of the surface. A near-surface layer zone was observed that extends for 100 µm or more where the composite has been altered from the bulk morphology. For longer exposure durations, it is evident that an environment barrier coating is necessary. The Mo-Si-B-based coating was thus evaluated for such an application.
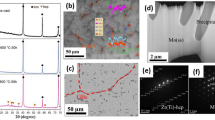
As an initial evaluation of the coating’s protective capability to an extreme arc jet environment, the Mo-Si-B coating was applied to pure Mo substrates. After multiple 15- to 20-min arc jet exposures, with temperatures reaching 1600 °C(see Ref 30 for the details of the arc jet exposure), the samples maintained their morphology, with no significant change in the coating microstructure as shown in Fig. 9(a). To further extend the study, the Mo-Si-B coating was applied to a ZrB2-SiC composite by the previously outlined two-step method. Again, after arc jet testing the sample demonstrated good coating adherence as shown in Fig. 9(b). Again, data suggest that the protection provided by the aluminoborosilica surface is related to retention of glass additives (i.e., Na, Al and B) that act to maintain a non-crystalline surface and a reduced oxygen partial pressure at the substrate/coating interface.

(a) SEM micrograph following multiple 15- to 20-min arc jet exposures at temperatures reaching 1600 °C (see (Ref 31) for the details of the arc jet exposure). The samples maintained their integrity, protecting the oxidation vulnerable Mo substrate. (b) SEM micrograph of the Mo-Si-B coating applied to a ZrB2-SiC composite after arc jet exposure. The sample exhibits good coating adherence and an unaltered substrate (adapted from Ref 29)
Interaction Between Mo-Si-B Coatings and CMAS
Thermal barrier coatings are highly developed material systems for increasing the durability and functioning of gas turbine engines (Ref 31, 32). However, one issue that has received recent attention by several groups is TBC degradation due to ingestion and infiltration of calcia-magnesia-alumino-silicate (CMAS) into the TBC structure (Ref 33, 34). Moreover, thermal barrier coatings are increasingly vulnerable to attack by molten CMAS sand. Thermal barrier coatings (TBCs) made of ZrO2 typically stabilized with 7 wt.% Y2O3 are used in gas turbine engines for aircraft engines, power generation and marine applications for higher operating engine temperatures. The molten CMAS attacks the TBC by infiltration into the coating and subsequently results in delamination cracks and damage to the substrate (Ref 35). In addition, the CMAS causes the TBC to exhibit extensive thermomechanical degradation during repeated thermal cycles which leads to a number of delamination mechanisms due to CTE mismatch between the TBC and CMAS. The CTE of TBC is relatively high (9-12 × 10−6 K−1) compared to that of CMAS (~3-4 × 10−6 K−1) (Ref 36, 37).
Only a few methods for alleviating CMAS attack on YSZ TBCs have been outlined in the literature. One protection method is to employ a sealant or a de-wetting outer layer on the TBC (Ref 31). However, as a result of thermal cycling during operation the outer layer was observed to crack and erode. Another technique is to apply an atmospheric plasma spray (APS) coating (Ref 25). Unfortunately, the morphology of the APS YSZ TBC is typified by the presence of microcracks in the TBC, porosity and “splat” boundaries. Consequently, the interaction between molten CMAS and TBC materials follows the corrosion mechanism noted above. In this current work, the interaction was analyzed between CMAS and MoSiB protective coating. The main idea of the experiment is to create a new type of protective coating as an effective source glass overlays that will interrupt the molten CMAS infiltration into thermal barrier coating by altering the chemical composition and the physical properties of the CMAS.
The MoSiB-based coating was applied to the Mo substrate samples by a pack cementation process where gas phase diffusion of Si and B reacts with the Mo surface to convert it into the coating. The pack cementation process was conducted at 1000 °C for 50 h. Following pack cementation, the surface is converted by conditioning into a mixture of MoSi2 and MoB. After pack cementation processing, the specimens were conditioned for 2 h at 1600 °C in ambient air.
In order to establish the interaction behavior of CMAS with the MoSiB-based coating, small quantities (12 mg/cm2) of CMAS powders were sprinkled on the top surface of the coated specimens (Ref 38). A synthetic CMAS paste was produced by a technique which is described by Kramer et al. (Ref 34). The CMAS-covered samples were subjected to oxidation exposure in air at 1200-1500 °C for 2-, 10- and 25-h exposures.
The results shown in Fig. 10 are representative of CMAS exposures. Initial contact of the CMAS with the coating results in the production of SiO2 and a limited amount of MoO2 by the coating. The MoO2 is incorporated into the CMAS to produce CaMoO4 oxide at the surface of the CMAS, as shown in Fig. 10(a). The layered structure after exposure in air these high temperatures is depicted schematically in Fig. 10(c). Upon exposure to Mo-Si-B coatings, the CMAS melts above a particular temperature and through its interaction with the MoO2 generated by the coating becomes depleted in calcium and crystallizes. The increase in the Al from the pack cementation process and the Ca depletion changes the CMAS composition to one that is easy to crystallize. Therefore, this process makes CMAS immobile and inhibits further interaction with the Mo-Si-B coating. This process occurs in addition to the excellent oxidation resistance at high temperatures demonstrated by the Mo-Si-B-based coating when applied to molybdenum alloys and several different substrates.

(a) Cross-sectional SEM micrograph of MoSiB coating after exposure for 25 h at 1200 °C in air, in contact with natural CMAS (Sand A). (b) Plan-view SEM micrograph of top surface of the CMAS oxidized for 25 h at 1400 °C in air showing the Ca-Mo oxide. (c) Schematic of the coating layer structure depicting the CMAS-MoSiB interactions (adapted from Ref 38)
Hot Corrosion Exposure
Hot corrosion is a phenomenon primarily encountered in gas turbine engines which operate in salt-containing environments. Salt from sea air can be ingested by an engine and form a stable melt in engine sections which are in the temperature span of 600-900 °C (Ref 39). Hot corrosion is a form of environmental attack in which a molten salt interacts with a material, causing accelerated corrosive attack. These interactions can lead to irregular and accelerated oxidation behavior. Fused Na2SO4 considered the dominant salt in hot corrosion can induce a fluxing of a normally protective oxide (Ref 40). Fuel impurities such as V can also contribute to the final salt scale chemistry. Multiple mechanisms have been put forward to attempt to explain the characteristics of hot corrosion attack, but of interest is the fluxing mechanism (Ref 41). Also of note is the equilibrium between the salt scale and the surrounding vapor environments. This determines the stability of the salt scale, in that still air promotes more stability of the melt, and moving air promotes the decomposition of the salt scale.
To evaluate the susceptibility of Mo-Si-B-based coatings to hot corrosion, the effects of fuel impurities and salt ingestion forming a molten salt scale on the surface of the coating must be considered. A solution of a 50/50 mixture by mass of Na2SO4 and NaVO3 (this mixture was selected because a salt scale containing V will tend to be acidic due to the acidic nature of V2O5, which results in the most severe attack (Ref 40, 41)) was employed to simulate the worst-case scenario. A slurry of the salt mixture was sprayed onto Mo-Si-B-coated Mo coins, and the solution was dried in a drying cabinet, depositing a film of salt. A Mo substrate was selected due to its lack of inherent oxidation protection at the exposure temperatures, so that only the performance of the coating is demonstrated. Salt-coated coins were then loaded into a furnace for cyclic exposure at 900, 1000 and 1300 °C. Due to the projected effect of vapor velocity, tests were carried out in both still and flowing air. Mass and morphology change was tracked between cycles. Finally, the possibility of vapor-phase attack was explored by exposing Mo-Si-B-coated coins without a salt scale, but instead a loose salt charge is loaded into the furnace upstream to expose the sample to salt vapor.
Compiled mass change data are presented in Fig. 11 for cyclic tests at 900 and 1000 °C for both the still and flowing air cases. Note the survival out to 21, 5-h cycles under flowing air conditions and the general increase in attack severity at higher temperature. Mass losses can be partially attributed to spallation of small fragments of the coating. Associated with this is a visual change in coating character.

Compiled mass change data for 900 and 1000 °C, still and flowing air, 50/50 Na2SO4/NaVO3 experiments. The uptick shown before the first cycle is the salt loading applied to the sample. Note the observed survival of a specimen out to 21, 5-h cycles with minimal losses for both the 900 and 1000 °C flowing air cases
The primary effect of hot corrosion attack on the coating is the crystallization of the amorphous silica scale into Cristobalite as demonstrated in the x-ray diffraction (XRD) scans presented in Fig. 12(a) and (b), which demonstrate this fact. Note the diffraction pattern consistent with Cristobalite in Fig. 12(b) replaces amorphous hump observed in Fig. 12(a). This transformation is rapid, as a single 5-h cycle is sufficient to eliminate any amorphous signal. Despite this transformation, the coating retains its protection of the vulnerable Mo substrate. The origin of the continued protection is revealed in the micrograph shown in Fig. 13. The image demonstrates the coating’s ability to self-heal and arrest cracks, by forming additional silica at the MoSi2-scale interface, slowing the progression of the oxidation.

(a) XRD scan of Mo-Si-B coating, pre-exposure. Note the strong amorphous signal at 22 degrees as well as the crystalline Mo5Si3 and MoSi2 phases present in the scan. (b) XRD scan of Mo-Si-B coating, after 3, 5 h cycles at 1000 °C. Note that the Mo5Si3 remains but the glassy signal and MoSi2 signatures are absent. This suggests a crystallization of vitreous silica to cristobalite

Secondary electron SEM micrographs of coated specimen subjected to hot corrosion. The specimen was exposed to 21 5 h cycles at 900 °C after loading 4.54 mg/cm2 salt mix. Note that despite cracking the coating has retained its protective ability for the vulnerable Mo substrate. New SiO2 formation acts to fill in the crack and acts as a self-healing mechanism. Furthermore, note that the cracks terminate at the Mo5SiB2 (T2) phase and do not continue into the substrate
Overall, the interaction between the Mo-Si-B coating structure and a molten salt scale results in near-total crystallization of the vitreous silica network. This transformation from silica to cristobalite causes incremental mass losses upon thermal cycling. This growth causes a plainly visible change in coating character. Mass losses are attributed to gradual spallation of the cristobalite, due to a significant volume change upon alpha-beta inversion at 270 °C (Ref 41). It is believed that this crystallization is catalyzed by the Na2O groups supplied by the salt facilitating transformation (Ref 41). Attack rates are enhanced when the gas composition is in equilibrium with the liquid scale, as this increases the temperature range of stable liquid. However, in the interior of a gas turbine engine such an equilibrium is not expected to be reached.
Conclusions
The robust natures of the Mo-Si-B-based coating which is effective in limiting oxygen penetration are derived through its ability to self-repair as needed by the regeneration of the outer scale by solid-state transformation of the coating from Si-rich to low-Si-rich phases. This self-regeneration process is especially important under cyclic thermal loading. The coating is highly capable of handling cyclic oxidation, since any cracking of the coating layer during cooling is repaired either through viscous flow of existing coating at sufficiently elevated temperatures, or through regenerative local production of additional borosilica. The T1 and T2 layers play a key role in the termination of crack propagation before it reaches the substrate, which would offer no self-protection and lead to catastrophic failure. The rapid generation of fresh borosilica at the crack walls serves not only to protect the coating from further oxidation, but also to provide an effective barrier to the CMAS and hot corrosion attack, preventing direct access to the coating or underlying substrate interface.
The use of plasma spraying of Molybdenum as the first step in the two-step Mo-Si-B coating method is effective in protecting different substrates such as SiC composites, ZrB2-SiC, Nb alloys and cermets from oxidation. By using plasma spraying to deposit Mo followed by a co-pack cementation of Si and B, the Mo-Si-B-based coating can be applied to provide enhanced oxidation protection to a wide range of substrates. This provides a proof-of-concept that the plasma spraying of Mo is an effective first step in generation of an oxidation-resistant Mo-Si-B-based coating. Plasma spraying the molybdenum enables the coating of larger and more complicated samples compared to the alternative carbonyl deposition method.
References
D.M. Dimiduk and J.H. Perepezko, Mo-Si-B Alloys: Developing a Revolutionary Turbine-Engine Material, MRS Bull., 2003, 28(9), p 639-645
J.H. Perepezko, The Hotter The Engine, The Better, Science, 2009, 326, p 1068-1069
J.A. Lemberg and R.O. Ritchie, Mo-Si-B Alloys for Ultrahigh-Temperature Structural Applications, Adv. Mater., 2012, 24(26), p 1-36
J.H. Perepezko and R. Sakidja, Oxidation Resistant Coatings for Ultra-High Temperature Refractory Mo-Base Alloys, Adv. Eng. Mater., 2009, 11, p 892-897
J.H. Perepezko and R. Sakidja, Extended Functionality of Environmentally-Resistant Mo-Si-B-Based Coatings, JOM, 2013, 65, p 307-317
E.J. Opila, N.S. Jacobson, D.L. Myers, and E.H. Copland, Predicting Oxide Stability in High-Temperature Water Vapor, JOM, 2006, 58(1), p 22-28
J.H. Perepezko, Surface Engineering of Mo-Base Alloys for Elevated Temperature Environmental Resistance, Annu. Rev. Mater. Res., 2015, 45, p 519-542
I. Spitsberg and J. Steibel, Thermal and Environmental Barrier Coatings for SiC/SiC CMCs in Aircraft Engine Applications, Int. J. Appl. Ceram. Technol., 2004, 1(4), p 291-301
P. Ritt, O.J. Lu-Steffes, R. Sakidja, J.H. Perepezko, W. Lenling, D. Crawmer, and J. Beske, Application of Plasma Spraying as a Precursor in the Synthesis of Oxidation-Resistant Coatings, J. Thermal Spray Tech., 2013, 22(6), p 992-1001
J.L. Sabourin and R.A. Yetter, High-Temperature Heterogeneous Reaction Kinetics of Tungsten Oxidation by CO(2), CO, and O(2), J. Propuls. Power, 2009, 25(2), p 490-498
J.H. Perepezko and R. Sakidja, Oxidation-Resistant Coatings for Ultra-High-Temperature Refractory Mo-Based Alloys, JOM, 2010, 62(10), p 13-19
P. Ritt, R. Sakidja, and J.H. Perepezko, Mo-Si-B Based Coating for Oxidation Protection of SiC-C Composites, Surf. Coat. Technol., 2012, 206(19-20), p 4166-4172
V.A. Ravi, S.D. Cramer, and B.S. Covino, Jr., Pack cementation coatings, ASM Handbook, Volume 13A: Corrosion: Fundamentals, Testing, and Protection, S.D. Cramer, and B.S. Covino, Jr., Eds., ASM International, Materials Park, 2003, p 763-771
R. Sakidja, F. Rioult, J. Werner, and J.H. Perepezko, Aluminum Pack Cementation of Mo-Si-B Alloys, Scr. Mater., 2006, 55, p 903
M. Akinc, M.K. Meyer, M.J. Kramer, A.J. Thom, J.J. Huebsch, and B. Cook, Mater. Sci. Eng., 1999, 261, p 16
M.K. Meyer, A.J. Thom, and M. Akinc, Oxide Scale Formation and Isothermal Oxidation Behavior of Mo-Si-B Intermetallics at 600-1000 °C, Intermetallics, 1999, 7, p 153
N.S. Jacobson, D.L. Myers, E.J. Opila, and E.H. Copland, Interactions of Water Vapor with Oxides at Elevated Temperatures, J. Phys. Chem. Solids, 2005, 66, p 471-478
E.J. Opila, Oxidation and Volatilization of Silica Formers in Water Vapor, J. Am. Ceram. Soc., 2003, 86, p 1238-1248
E.J. Opila, Paralinear Oxidation of CVD SiC in Water Vapor, J. Am. Ceram. Soc., 1997, 80, p 197-205
E.J. Opila, Paralinear Oxidation of Silicon Nitride in a Water-Vapor/Oxygen Environment, J. Am. Ceram. Soc., 2003, 86, p 1256-1261
F. Rebillat, X. Martin, E. Garitte, and A. Guette, Design, Development, and Applications of Engineering Ceramics and Composites, Am. Ceram. Soc., 2010, 215, p 151-166
K. Gente, F. Rebillat, and F. Langlais, High Temperature Corrosion and Materials Chemistry III: Proceedings of the International Symposium, Pennington NJ Electrochemical Society, 2003, pp. 545-556.
P. Mandal, A.J. Thom, M.J. Kramer, V. Behrani, and M. Akinc, Mater. Sci. Eng. A, 2004, 371, p 335-342
F.A. Rioult, S.D. Imhoff, R. Sakidja, and J.H. Perepezko, Transient Oxidation of Mo-Si-B Alloys: Effect of the Microstructure Size Scale, Acta Mater., 2009, 57, p 4600-4613
J.M. Drexler, K. Shinoda, A.L. Ortiz, D.S. Li, A.L. Vasiliev, A.D. Gledhill, S. Sampath, and N.P. Padture, Air-Plasma-Sprayed Thermal Barrier Coatings that are Resistant to High-Temperature Attack by Glassy Deposits, Acta Mater., 2010, 58, p 6835-6844
T.A. Jackson, D.R. Eklund, and A.J. Fink, High Speed Propulsion: Performance Advantage of Advanced Materials, J. Mater. Sci., 2004, 39, p 5905
W.G. Fahrenholtz and G.E. Hilmas, Oxidation of Ultra-High Temperature Transition Metal Diboride Ceramics, Int. Mater. Rev., 2012, 57, p 61
M.M. Opeka, I.G. Talmy, and J.A. Zaykoski, Oxidation-Based Materials Selection for 2000 C+ Hypersonic Aerosurfaces: Theoretical Considerations and Historical Experience, J. Mater. Sci., 2004, 39, p 5887
E. Eakins, D.D. Jayaseelan, and W.E. Lee, Toward Oxidation-Resistant ZrB2-SiC Ultra High Temperature Ceramics, Metall. Mater. Trans. A, 2011, 42A, p 878
P.J. Ritt, P.A. Williams, S.C. Splinter, and J.H. Perepezko, Arc Jet Testing and Evaluation of Mo-Si-B Coated Mo and SiC-ZrB2 Ceramics, J. Eur. Ceram. Soc., 2013, 34(15), p 3521-3533
N.P. Padture, M. Gell, and E.H. Jordan, Thermal Barrier Coatings for Gas-Turbine Engine Applications, Science, 2002, 296, p 280-284
R.C. Reed, Superalloys and Coatings for High-Temperature Applications, JOM, 2006, 58, p 36
X. Chen, Calcium-Magnesium-Alumina-Silicate (CMAS) Delamination Mechanisms in EB-PVD Thermal Barrier Coatings, Surf. Coat. Technol., 2006, 200, p 3418-3427
S. Kramer, J. Yang, C.G. Levi, and C.A. Johnson, Thermochemical Interaction of Thermal Barrier Coatings with Molten CaO-MgO-Al2O3-SiO2 (CMAS) Deposits, J. Am. Ceram. Soc., 2006, 89, p 3167-3175
B.J. Harder, J. Ramirez-Rico, J.D. Almer, K.N. Lee, and K.T. Faber, Chemical and Mechanical Consequences of Environmental Barrier Coating Exposure to Calcium-Magnesium-Aluminosilicate, J. Am. Ceram. Soc., 2011, 94, p S178-S185
A.G. Evans and J.W. Hutchinson, The Mechanics of Coating Delamination in Thermal Gradients, Surf. Coat. Technol., 2007, 201, p 7905-7916
S. Kramer, S. Faulhaber, M. Chambers, D.R. Clarke, C.G. Levi, J.W. Hutchinson, and A.G. Evans, Mechanisms of Cracking and Delamination Within Thick Thermal Barrier Systems in Aero-Engines Subject to Calcium-Magnesium-Alumino-Silicate (CMAS) Penetration, Mater. Sci. Eng. A, 2008, 490, p 26-35
I.P. Downs, J.H. Perepezko, R. Sakidja, and S.R. Choi, Suppressing CMAS Attack with a MoSiB-Based Coating, Surf. Coat. Technol., 2014, 239, p 138-146
R.A. Rapp, Hot Corrosion of Materials: A Fluxing Mechanism?, Corros. Sci., 2002, 44(2), p 209-221
N. Eliaz, G. Shemesh, and R.M. Latanision, Hot Corrosion in Gas Turbine Components, Eng. Fail. Anal., 2002, 9(1), p 31-43
M.G. Lawson, H.R. Kim, F.S. Pettit, and J.R. Blachere, Hot Corrosion of Silica, J. Am. Ceram. Soc., 1990, 73(4), p 989-995
A. Lange, M. Heilmaier, T. Sossamann, and J.H. Perepezko, Oxidation Behavior of Pack-Cemented Si-B Oxidation Protection Coatings for Mo-Si-B Alloys at 1300 °C, Surf. Coat. Technol., 2015, 266, p 57-63
Acknowledgements
The support from the Office of Naval Research (N00014-10-1-0913 and N00014-17-1-2575, Dr. David A. Shifler (program manager)) is most gratefully acknowledged. The continued interest and encouragement from Dr. Steven G. Fishman is appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Perepezko, J.H., Sossaman, T.A. & Taylor, M. Environmentally Resistant Mo-Si-B-Based Coatings. J Therm Spray Tech 26, 929–940 (2017). https://doi.org/10.1007/s11666-017-0565-2
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11666-017-0565-2