
Abstract
The principal chemical components in iron ore sintering are Fe2O3, CaO, and SiO2. This sintering process consists of three key steps: heating, holding at peak temperature, and cooling. During the cooling stage, a liquid oxide solidifies to form the final sinter microstructures. To investigate the fundamental processes taking place during the cooling of sinters, a new experimental technique has been developed that allows the stages of solidification to be determined in isolation, rather than inferred from the final microstructures. Fe2O3-CaO-SiO2 oxide samples of a bulk composition having a CaO/SiO2 mass ratio of 3.46 and 73.2 wt pct Fe2O3 were cooled in air from 1623 K (1350 °C) at 2 K/s, quenched at 5 K temperature intervals from 1533 K to 1453 K (1260 °C to 1180 °C), and analyzed using Electron Probe Micro X-Ray Analysis (EPMA). During cooling, four distinct stages were observed, consisting of the phase assemblages Liquid + Hematite (I), Liquid + Hematite + C2S(II), Liquid + C2S + CF2(III), and C2S + CF2 + CF (IV). This solidification sequence differs from that predicted under equilibrium and Scheil–Gulliver Cooling. Importantly, no Silico-Ferrite of Calcium (SFC) phase was observed to form on solidification of the liquid. Based on the microstructures formed and liquid compositions, measured by EPMA, it was demonstrated that kinetic factors play a major role in determining the phases and microstructures formed under the conditions investigated.
Similar content being viewed by others


Introduction
Over one billion tons of steel are produced annually using the iron blast furnace/basic oxygen steelmaking route. The sintering process is a key process step in the preparation of a competent feed from iron ore fines for the iron blast furnace, with some furnaces operated almost entirely on sinter.[1] In the sintering process the iron oxide fines, flux, coke and return sinter are heated and partly melted, to produce bonded multi-phase particles. The use of sinter in the furnace feed increases the productivity of iron blast furnaces[2] and enables a wide range of low cost ore feed and recycled materials to be processed.
It was suggested[3] and later confirmed,[4,5,6] that the phases present and microstructures in sinter influence their strength and reducibility, and in turn the efficiency and productivity of the iron blast furnace. For this reason, there are ongoing efforts to identify optimal sinter microstructures and how they are produced.
It has been observed[6,7,8,9] that there are three key stages in the sintering process: heating, peak temperature, and cooling. Solid state reactions have been observed to occur on heating, with new phases and microstructures being formed in the oxide material. As the peak temperature is approached, some of these phases melt or dissolve. During cooling, the liquid solidifies to form new crystalline and amorphous phases. Given the complexity of the sintering processes, understanding and controlling the processes to obtain the desired microstructures and properties presents an ongoing technical and scientific challenge.
The principal elements present in iron ore sinters are Fe-Ca-Si-Al-Mg-O; iron can be in the form of ferric or ferrous iron depending on the temperature and oxygen partial pressure. There is a lack of information available for the range of compositions and temperatures observed in iron ore sinters.
Pilot scale sinter pot tests are undertaken routinely as a form of quality control to determine the effects of changing ore blends and key process variables.[10] This approach whilst reproducing industrial sintering conditions does not explain why or how particular phases and microstructures are formed.
Laboratory based techniques used to investigate iron ore sintering have included the use of infrared heating furnaces,[6,7,8] electrically heated tube furnaces,[11,12] and in-situ X-ray powder diffraction (XRD).[13,14,15,16,17,18] Of these techniques, the infrared furnaces and in-situ XRD provide the most control over the heating and cooling processes. The infrared furnace technique allows samples to be prepared at heating and cooling rates representative of sintering conditions, but using this approach it is only possible to observe the initial and final states of the mixtures.[6,7,8] The in-situ XRD technique enables the phases and the temperature at which they form to be determined, but it is not possible to heat and cool samples at rates representative of sintering conditions and does not provide microstructural information.[13,14,15,16,17,18]
These previous studies have demonstrated that there are many factors that influence the outcomes of the sintering process. These factors include material characteristics, such as feed composition and mineralogy, and process characteristics, such as coke addition and draft suction. The large number of process variables is such that developing empirically based models to predict product microstructure presents an extremely difficult if not intractable problem.
A very different and fundamentally based approach is proposed by the authors based on the hypothesis that the principal phases and microstructures are formed in sinter during the solidification of the oxide liquids. A series of studies have been undertaken to demonstrate the validity of this view, these include;
-
Establishing the phase equilibria, and in particular the liquidus surface, in selected oxide systems
-
Systematic studies of the solidification processes taking place as a function of key process variables
As part of the first stage of this approach, experimental techniques have been developed[19] to accurately measure the solidus and liquidus compositions in these low-silica liquids. These techniques have been applied to the characterisation of phase equilibria in selected oxides systems relevant to iron ore sintering. The Fe2O3-rich corner of the “Fe2O3”-CaO-SiO2 system in air is the simplest ternary oxide system that can be used to represent iron ore sintering. Following earlier studies,[20] the liquidus surface and the primary phase field for the SFC (silico-ferrite of calcium) solid solution have now been established.[19,21] This provides the solid basis for further systematic studies to be undertaken to understand the influence of kinetic processes on the resulting sinter microstructures in this system.
The second stage involves the development of suitable experimental techniques that enable the phases formed on solidification, the sequence of formation, and the resulting microstructures to be characterized under known and controllable process conditions. This is the focus of the present article.
Experimental Technique
An improved technique has been developed to enable the determination of the processes occurring during the controlled cooling of synthetic sinter. Unlike previously used techniques,[6,7,8,11,13,14,15,16,17,18] the improved approach enables samples to be quenched from any temperature during the solidification sequence, enabling the phases and liquid present at temperature to be determined. This allows each of the processes taking place during solidification to be observed in isolation, separate from the processes occurring during heating. The ability to rapidly quench the sample and to accurately capture the microstructures and phase compositions present at temperature are critical to the experimental design. The controlled cooling rates investigated with the improved experimental technique are within the range measured in industrial iron ore sintering.[22]
Synthetic sinters have been prepared from mixtures of high purity oxide powders of Fe2O3 (> 99.5 wt pct, Alfa Aesar), CaCO3 (> 99.95 wt pct, Alfa Aesar), and SiO2 (> 99.9 wt pct, Alfa Aesar). Before mixing, all the oxides were calcined in air to remove absorbed and chemically bonded water, and decarbonate CaCO3. CaCO3 was heated above 1173 K (900 °C) for at least 5 hour, and the SiO2 and Fe2O3 were pre-heated above 673 K (400 °C) for 4 hour. The masses of the individual powders were weighed on a digital balance to a precision of 1 wt pct to obtain the target mass. The powders were mixed with an agate mortar and pestle to achieved intimate contact. The mixed powders are pressed by a hydraulic press in a tool steel die into a pellet (0.1 g, 6 mm ø). The oxide samples are supported on a Pt-Rh (70 wt pct Pt, 30 wt pct Rh) wire (0.5 mm ø) spiral. The support is as small as that practically achievable, typically with a diameter slightly larger than that of the pellet. The small sample remains attached to the substrate on cooling due to surface tension forces.
The oxide pellet and wire support are heated and cooled in air in an electrically heated vertical tube furnace (alumina reaction tube, 38mm OD, 30mm ID), with lanthanum chromite heating elements. The peak temperature and the temperature profile within the furnace (Pyrox) are maintained at constant values using a feedback controller (Eurotherm 2404) and a Pt/Rh controller thermocouple positioned immediately adjacent to the uniform hot zone of the furnace alumina tube.
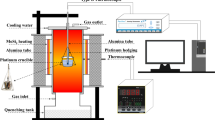
The sample is vertically suspended within the reaction tube in the furnace from an alumina rod, see Figure 1. The vertical position of the alumina rod, and in turn the sample, is controlled with a positioning motor affixed to the top of the furnace. The temperature of the sample is measured by a separate specimen thermocouple (B type, 70 pctPt/30 pctRh-94 pctPt/6 pctRh) positioned approximately 5 mm above the sample. This thermocouple cannot be immersed in the slag since to do so would destroy the alumina rod and in turn the thermocouple. The accuracy of the thermocouple reading is estimated to be within 3 °C of the standard thermocouple, calibrated by the Australian Government National Measurement Institute, within the temperature range investigated.

Schematic diagram of the experimental apparatus for the controlled cooling of oxide liquids
In preliminary studies using the above technique, it was shown that differences between the measured and actual sample temperature can result from changes in sample position, sample size, and solidification processes. Since the sample is positioned approximately 5 mm lower than the specimen thermocouple tip, during cooling it is in a lower temperature region of the furnace than the specimen thermocouple tip. Measurements made during cooling with and without a sample present showed that the presence of a sample leads to a higher temperature measurement by the thermocouple tip. This difference is believed to be due to the thermal mass of the sample and the occurrence of exothermic processes (i.e., heat of transformation and heat of reaction) during cooling, resulting in a higher specimen temperature and the transfer of heat from the sample to the thermocouple tip. Due to these processes, the specimen temperature approaches that measured by the thermocouple tip. For the conditions selected for these experiments, this difference is estimated based on the temperature profile within the furnace tube, for temperatures above 1173 K, to be within 5 K. The results presented hereafter refer to the temperature measured by the thermocouple.
The experiment is set up by first setting the peak temperature in the furnace hot zone and allowing the temperature profile to come to a pseudo-steady state condition. The sample is cooled at a controlled rate by moving the sample and thermocouple down through the hot zone of the furnace. The rate of movement, and hence the cooling rate, is controlled by programmable computer software, written specifically for this purpose.
To perform a controlled cooling, the programmable computer software is first calibrated. The calibration is performed by moving the alumina rod supporting the sample at a constant velocity (0.5 mm/s) through the tube furnace. This provides the software with a temperature profile able to predict the temperature at each position in the furnace during cooling.
The temperature profile of the furnace is used by the computer software to calculate the change in position with time required to achieve the target constant cooling rate (K/s). Once calibrated and the position variation with time is calculated by the software, the sample is able to be cooled at a constant rate. The sample is moved through the furnace at the calculated rate by the positioning motor, controlled by the programmable computer software. This system is able to achieve a cooling rate within 10 pct of the target value.
To perform an experiment, the alumina support tube is lowered to the bottom of the reaction tube, the sample is attached to the tube with the platinum wire. The moving stage is raised to the hot zone of the furnace, and the sample is held for four minutes at or above 1623 K (1350 °C) for melting and homogenisation. After homogenisation, the sample is lowered to achieve a constant cooling rate. On reaching the target temperature the sample is quenched in water by releasing the sample, which falls under gravity into the quenching medium. Quenched samples are washed briefly in water and air dried prior to mounting in epoxy resin. The samples are polished with conventional metallographic techniques, using the Struers system. Samples were first ground with SiC paper to section the sample, followed by the polishing. The first stages of polishing were performed with diamond suspended in water (9 μm) and the last two stages were with diamond paste (3 μm, 1 μm) suspended in kerosene. The later procedure is undertaken to ensure that phases that are reactive with water in the microstructure are not damaged or removed during sample preparation.
The sample cross-sections were characterized, initially using optical microscopy for phase and microstructure identification using a reflected light microscope (Olympus Provis AX70TRF). For electron microscope analysis, the polished surfaces were carbon coated using a carbon coater (Quorum Technologies, QT150TES). For high resolution imaging, a field emission scanning electron microscope (FE-SEM) (JEOL 7001F; Japan Electron Optics Ltd., Tokyo, Japan) was used with an accelerating voltage of 15 keV in backscatter mode.
Phase compositions were measured by an electron probe X-ray microanalysis (EPMA) technique with wavelength dispersive spectroscopy (WDS) (JEOL 8200L EPMA; Japan Electron Optics Ltd., Tokyo, Japan). An electron beam generated from a tungsten filament was generated at a voltage of 15 keV and current of 20 nA. The standard Duncumb–Philibert atomic number, absorption, and fluorescence correction (ZAF-correction) supplied with the probe was used. Selected point analysis of phases within the sample was performed. Three standards were used for calibration, pure CaSiO3, Fe2O3, and Al2O3 (all standards from the Charles M. Taylor Co., Stanford, CA). The accuracy of the composition measurements was estimated to be within 1 pct of the measured value. The proportion of elements in different oxidation states were not measured; the iron in the sample was assumed to be in the Fe3+ state for presentation purposes.
The experimental conditions were selected to reflect conditions in industrial processes, provide controllable and repeatable results, and to investigate the processes of interest to industrial sinter using the phase equilibria data available for the “Fe2O3”-CaO-SiO2 system in air.[19] The bulk composition of the samples was selected such that the liquid was in the hematite primary phase field, fully molten at the peak temperature and forms the silico-ferrite of calcium (SFC) phase on equilibrium cooling. The fully molten state at the peak temperature ensures uniformity in the starting material and that all crystalline phases are only formed during cooling, providing reproducibility and control.
The bulk composition of the oxide mixture, 72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO (CaO/SiO2 ratio of 3.46 wt/wt), was selected as this was predicted to form the SFC, Fe2O3, and C2S phases during equilibrium cooling of the liquid in air.[19] The liquidus temperature is estimated to be 1568 K (1295 °C). A constant cooling rate of 2 K/s was selected as this is typical of the cooling rate in industrial processes and reduces the number of variables in the experiments.[22]
In a sinter bed, there is both macroscale and microscale heat transfer. The aim of the experimental work is to replicate the microscale processes, and as such, the microscale heat transfer requires replication with the experimental technique. In the iron ore downdraft sinter process,[23] the combined heat and combustion fronts move progressively down through the sinter bed. This implies 1D directional heating and solidification is taking place however this is a packed bed made up of individual granules. Heat transfer around and within individual granules should be taken into account.
The Biot number[24] for sinter particle cooling in industrial sinter machines was calculated, using the following assumptions;
-
Temperature of air—473 K (200 °C)
-
Temperature of sinter—1573 K (1300 °C)
-
Thermal conductivity of solids—2 W/m K
-
Gas velocity—2 m/s
-
Sinter particle diameter—0.02 m
Using these parameters, and using the heat transfer correlations for both forced and natural convection around a spherical particle,[24] the Biot number was found to be less than 0.1. Thus indicating that the rate of heat conduction within the particles is greater than the rate of heat transfer by forced convection from the surrounding gas phase to the particles surface. As such, it can be concluded that the temperature within individual sinter particles is relatively uniform and constant, resulting in volumetric solidification rather than directional solidification. The experimental technique is thus designed to reproduce the volumetric solidification by selecting appropriate experimental conditions.
Initial tests were performed to confirm that the selected conditions allowed for liquid formation and homogenisation prior to solidification. Samples were held at 1623 K (1350 °C) for 4 minutes, and then quenched to room temperature. In the resulting structure the composition of the sample, as measured by EPMA, was found to be uniform across the whole sample cross section. No primary phase solidification crystals were resolvable using the electron microscope. The uniform composition indicates the quenched matrix was either amorphous or microcrystalline in structure. These observations indicate that, under the conditions tested, the sample was fully liquid and of homogeneous composition at the peak temperature, which was the start of the controlled cooling process.
In those cases in which a high quenching rate has not been achieved in the sample, the presence of fine crystals can be observed in the material that is ostensively liquid at the temperature from which it was quenched. The difference in the size of the crystals, and the phases that are present, are used to differentiate these regions from the principal sequence of crystal formation that has taken place during the controlled cooling step.
Preliminary tests were performed to determine the conditions that enable volumetric solidification to occur within the samples. Samples sizes between 0.1 and 0.2 g were tested and they were found to have a uniform microstructure with position within the sample, indicating that this sample size range provides a uniform composition and temperature profile during cooling. Samples with higher masses, e.g., 0.4 g, were found to have an uneven distribution of microstructures within the solidified samples. These preliminary tests were repeated multiple times to confirm that these conditions are able to produce reproducible results and a uniform temperature profile within the sample. For this reason, a sample size of 0.1 g, with an spheroid diameter of 0.4 mm and height of 0.3 mm once molten, as measured from quenched samples, was selected for the main study.
Theoretical Solidification Sequences
Equilibrium Cooling
Equilibrium cooling assumes infinitely rapid mass transfer in all phases and infinitely fast reaction kinetics. This results in equilibrium phases and phase assemblages forming at all temperatures during the cooling process. Under equilibrium cooling of the liquid, with the bulk composition (72.7 wt pct Fe2O3, 6.1 wt pct SiO2, and 21.1 wt pct CaO) shown as X in Figure 2, from 1623 K (1350 °C) to below 1423 K (1150 °C) five stages of solidification occur. The liquidus of the selected composition is approximately 1548 K (1275 °C). The first stage is the nucleation and growth of the primary phase, hematite, from the liquid (L → Fe2O3). At the conclusion of this stage, the maximum proportion of hematite present in the system is approximately 10 wt pct. Stages 2 and 3 involve the formation of SFC. During stage 2, SFC is formed though the peritectic reaction between the liquid and hematite (L + Fe2O3 → L + SFC). The peritectic reaction occurs over a small temperature range, over which the liquid composition follows the Hematite-SFC boundary curve away from the saddle point (d) towards the CF2-SFC-Hematite peritectic point (a) (approximately 1225 °C, 1498 K). The SFC phase is a solid solution with the end members CF3-C4S3, with the composition of this phase in equilibrium with liquid changing during cooling.[19] The reaction proceeds to completion such that no hematite remains at temperatures below approximately 1529 K, (~ 1256 °C). On further cooling during stage 3, the SFC continues to form (L → SFC) with the liquid composition moving across the SFC primary phase field. Stage 4 starts at approximately 1508 K (1235 °C) with the formation and precipitation of C2S together with SFC (L → SFC + C2S). Stage 5 occurs with the SFC reacting with the liquid at 1489 K (1216 °C) to form C2S and CF2 (L + SFC → C2S + CF2) until all the liquid is transformed. The phases present after this final reaction is complete are SFC, C2S and CF2.

Adapted from Ref. [19]
Predicted liquid compositions on equilibrium cooling of bulk composition X (72.74 wt pct Fe2O3, 6.11 wt pct SiO2 and 21.14 wt pct CaO) from 1623 K to 1453 K (1350 °C to 1180 °C) in the “Fe2O3”-CaO-SiO2 system in air
Scheil–Gulliver Cooling
The predicted Scheil–Gulliver cooling is different from equilibrium cooling and provides a second point of comparison for the observed processes. This cooling process assumes that solid state reactions, peritectic reactions and solid phase diffusion do not occur, diffusion in the liquid is rapid and solidification reactions achieve equilibrium. This results in solid phases, once formed, remaining and not changing in composition with further solidification or the formation of other phases. Under Scheil–Gulliver cooling, for the same bulk composition (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO), indicated as X in Figure 3, and temperature range (1623 K to 1423 K, 1350 °C to 1150 °C), five processes occur (see Figure 3). The liquidus of this liquid under Sheil-Gulliver cooling is the same as equilibrium cooling, 1548 K (1275 °C). The first stage is the nucleation and growth of stoichiometric primary phase hematite from the liquid (L → Fe2O3). The second stage of solidification occurs with the solidification of the SFC phase (L → SFC), starting at 1529 K (1256 °C). Unlike equilibrium cooling, the hematite does not react with the liquid and remains present during the solidification of SFC, contributing to approximately 10 wt pct of the total mass. Stage 3 occurs with SFC and C2S solidifying concurrently (L → SFC + C2S), starting at approximately 1508 K (1235 °C). The formation of SFC under Sheil-Gulliver cooling is characterized by coring, with the composition in the SFC solid solution changing during cooling. Stage 4 occurs with the concurrent formation of C2S and CF2 (L → C2S + CF2), starting at 1489 K (1216 °C). The fifth and final solidification process is the formation of the ternary eutectic C2S, CF2, and CF (L → C2S + CF2 + CF) at 1465 K (1192 °C). The final phase assemblage under this non-equilibrium cooling process consists of hematite (Fe2O3), SFC, C2S, CF2, and CF. This differs from equilibrium cooling as the peritectic reactions, the redissolution of hematite (L + H → SFC) and SFC (L + SFC → CF2), do not occur and as a result hematite remains at low temperature and CF forms during the final stage of solidification.

Adapted from Ref. [19]
Predicted liquid composition on cooling of the bulk composition X (72.74 wt pct Fe2O3, 6.11 wt pct SiO2 and 21.14 wt pct CaO) from 1623 K to 1453 K (1350 °C to 1180 °C) under Sheil-Gulliver cooling in the “Fe2O3”-CaO-SiO2 system in air
Results
The initial experiments were performed with samples cooled at 2 K/s from fully liquid conditions at 1523 K to 1073 K (1350 °C to 800 °C). Within this range, complete solidification was found to have taken place and there was no residual liquid at 1073 K (800 °C). An example of the typical microstructure formed under these conditions is given in Figure 4.

Example of the complex microstructure formed on cooling from 1623 K to 1073 K (1350 °C to 800 °C), at 2 K/s in air (72.74 wt pct Fe2O3, 6.11 wt pct SiO2 and 21.14 wt pct CaO). (H hematite, C2S 2CaO·SiO2, CF2 CaO·2Fe2O3, CF CaO·Fe2O3). (b) Detail of (a) (Backscattered electron (BSE) micrographs)
Using FE-SEM and EPMA measurements, the phases present in the sample were identified to be hematite (H, Fe2O3), dicalcium Silicate (C2S, 2CaO.SiO2), calcium ferrite (CF, CaO·Fe2O3), and calcium diferrite (CF2, CaO·2Fe2O3). Notably, the SFC phase was absent in all the samples examined. The preliminary analysis of the phases and microstructures demonstrated that those present were different from those anticipated for both equilibrium and Scheil–Gulliver cooling.
To provide a clear understanding of the sequence of reactions, a series of experiments were performed. These experiments were undertaken with samples cooled from the same starting temperature (1350 °C), and the same cooling rate (2 K); samples were quenched from selected temperatures between 1543 K and 1453 K (1270 °C and 1180 °C).
There are significant experimental difficulties in preparing and characterizing the samples that have been cooled under controlled conditions to these temperatures; very high cooling rates are required to retain the liquid as amorphous or microcrystalline phase on quenching. The high proportion of solids present provide sites for heterogeneous nucleation and growth of new and existing phases on quenching of the sample. Care must be exercised to ensure that structures formed on quenching are not interpreted as part of the controlled cooling sequence.
Between a fully homogeneous liquid and the final microstructure after complete solidification, four intermediate stages of solidification were observed in these experiments, each resulting in the formation of specific phases and phase assemblages. Within a single sample, multiple assemblages were able to be observed for a range of temperatures. The formation of new phases did not occur simultaneous in all regions of the sample at a single temperature, suggesting that nucleation occurred independently in these regions. The four, in the order of formation, are as follows, Liquid + H (Assemblage I), Liquid + H + C2S (Assemblage II), Liquid + C2S + calcium diferrite (Assemblage III), and finally C2S + calcium diferrite + calcium ferrite (Assemblage IV). Although the resultant total phase assemblage is additive, consisting of all solids that formed in the previous stages, for ease of description of the solidification phenomena, each of the phase assemblages are presented as occurring independently of the pre-existing solids. The temperatures at which the different phase assemblages were observed are illustrated in Figure 5.

Phase assemblages observed at temperature during liquid solidification (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO)
L + H (Assemblage I)
Assemblage I, involving the formation of hematite crystals, was the first phase assemblage observed during cooling from 1533 K to 1528 K (1260 °C to 1225 °C). Figure 6 shows a micrograph illustrating the typical hematite primary phase and microstructure.

Backscattered electron micrograph showing the locations of liquid compositions able to be measured Assemblage I, samples cooled from 1623 K to 1518 K (1350°C to 1245°C) at 2 K/s in air (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO). Line 1—close to hematite and a distance from the sample surface, Line 2—far from the hematite and close to the sample surface (H hematite, L liquid, Pt platinum)
The hematite was observed to be in the form of large, faceted dendritic (skeletal) crystals (>100 μm long, ~ 30 μm wide), distributed throughout the liquid; a larger proportion of solids was observed to be present near the platinum substrate. The hematite crystals increased in size and number with decreasing temperature. At the highest temperatures, just below the liquidus, these crystals were only observed in association with the substrate.
The composition of the liquid perpendicular to a planar hematite crystal was measured by point analysis with EPMA. The concentration profile is illustrated in Figure 7 with data from Table II, which shows the liquid composition as a function of distance from hematite. The composition profile was measured with the composition measured as close to the melt-hematite interface as possible without the excitation volume encompassing both melt and hematite.

Liquid composition as a function of distance from the hematite-liquid interface. Sample prepared by cooling a homogenous liquid from 1623 K to 1543 K (1350 °C to 1270 °C) at 2 K/s, followed by quenching to room temperature (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO). The melt composition was measured as close as possible to the hematite without the excitation volume intersecting the hematite
The liquid composition was found to vary with distance from the hematite/liquid interface. The composition of the liquid remote from the hematite found to have a Fe2O3 concentration close to that predicted by the liquidus surface[19] between hematite and liquid (i.e., the hematite and liquid equilibrium) at the temperature from which it was quenched. The liquid close to the hematite surface was observed to be poor in Fe2O3 relative to the high temperature equilibrium.[19] This indicated that the concentration profiles observed are a result of crystal growth during the quenching processes rather than present during crystal growth during the controlled cooling.
It was observed that the liquid composition was closest to equilibrium when measured at least 40 μm from hematite and within 40 μm of the surface of the sample. The liquid composition close to the sample surface and far from hematite was measured to have typically 5 wt pct more Fe2O3 than the other regions of the sample. This is illustrated in Figure 6, where Line 2 was measured to have an average Fe2O3 concentration of 70.7 wt pct and Line 1 with 65.2 wt pct. On quenching, the surface of the sample is the first to solidify while the center of the sample was still molten, allowing a short time for mass transfer to the liquid in the center of the sample and for further solidification of hematite to take place.
Liquid + H + C2S (Assemblage II)
Assemblage II was observed to form between 1508 K and 1473 K (1235 °C and 1200 °C) and was the second phase assemblage observed on cooling following the formation of the initial hematite dendrites. Figure 8 shows typical micrographs illustrating this phase assemblage.

Example of typical microstructures observed in Assemblage II, (a) sample cooled from 1623 K to 1498 K (1350 °C to 1225 °C) at 2 K/s in air (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO), (b) detail of microstructures in (a) (BSE) (H hematite, L liquid)
In this phase assemblage, the C2S phase was observed to be present as both individual crystals and in a coupled microstructure with hematite. The different microstructures were either formed concurrently or separately as cooling occurred. The C2S/liquid interface in all cases was non-faceted.
The C2S was observed as a thin layer on the primary hematite (IIa), individual C2S dendrites (IIb), and a coupled H + C2S microstructure (IIc). The layer of C2S on hematite (IIa) was observed on most of the hematite crystals, and was only 2 to 3 micrometers in thickness.
The coupled microstructure (H-C2S) (IIc) was found to surround all the individually formed hematite crystals, forming on either the hematite or the thin layer of C2S. In the coupled microstructure, hematite is the continuous phase and C2S is present as either rods or as an irregular unfaceted microstructure. The phase ratio is approximately 2 H:1 C2S by volume. The volume fraction of this coupled microstructure increased with decreasing temperature.
The individual C2S (IIb) appeared as unfaceted crystals either isolated in the liquid, or dendritic structures surrounded by either the coupled (H-C2S) microstructure or the liquid. Analysis of the 2D microstructure indicates that the individual and isolated C2S crystals are in fact part of the dendritic structures. The individual crystals were not observed in all regions of assemblage II, increasing in proportion and occurrence with decreasing temperature. These individual crystals appear to occasionally split to form two main branches. The crystals themselves are smaller than the hematite, and are observed to be up to 10 μm in width and 50 μm in length. The isolated crystals are not directly associated with the hematite, with only a Liquid/C2S interface observed. The crystals themselves are larger than the C2S crystals in the coupled microstructure.
L + C2S + CF2 (Assemblage III)
Phase assemblage III was observed to form between 1215 K and 1473 K (1215 and 1200 °C). Figure 9 shows a micrograph illustrating this phase assemblage. The high proportion of solids present in this temperature interval lead to difficulties in retaining liquid on quenching the sample. An increase in the proportion of solids increased the area of surfaces on which further solidification and nucleation is able to occur on quenching.

Microstructures observed in Assemblage III, samples cooled from 1623 K to 1488 K (1350 °C to 1215 °C) at 2 K/s in air (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO). (H hematite, C2S dicalcium silicate, L liquid) (b), (c) detail of (a) (BSE)
The microstructure observed consists of individual CF2 crystals and a coupled C2S-CF2 microstructure. The individual CF2 crystals were observed to form on the hematite and the coupled C2S-CF2 microstructure on the individual CF2 crystals. No change in crystal size or shape, or reduction in volume fraction of hematite primary phase crystals. This indicates no measurable dissolution of previously solidified hematite has taken place during this stage.
The individual CF2 crystals (IIIa) were observed to form thin faceted needles or plates (up to ~ 80 μm long and 5 μm wide), with an interface with both hematite, liquid and occasionally C2S. Some of the individual CF2 crystals were curved, providing an indication of the direction of growth or solidification. The individual crystals were observed to be thin and formed splitting tips. The splitting tips potentially indicated that the CF2 formed as a series of parallel plates, resulting in the appearance as an individual needle or plate. The length of the crystals appeared to be physically limited by the presence of solids formed following solidification at higher temperature. The smallest crystals were observed to form an interface with one solid phase, Hematite. The CF2 crystals were observed to have similar orientations and associations with other solids, but differed in size. The CF2 crystals were observed to form around the individual C2S crystals.
The coupled C2S-CF2 microstructure (IIIb) was only observed in some regions of assemblage III. The coupled microstructure is illustrated with Figure 10, as this microstructure is more clearly illustrated in Assemblage IV. The CF2 formed the continuous phase and C2S as rods (< 1 μm diameter), indicating that CF2 formed a larger volume proportion of the microstructure. The coupled microstructure was always located in association with the individual CF2 crystals, forming both in close proximity to and far from the solidified hematite. At the interface with the liquid, the C2S formed ahead of the CF2 with an unfaceted interface while the CF2 formed a faceted interface.

Microstructures observed to form in Assemblage IV, samples cooled from 1623 K to 1073 K (1350 °C to 800 °C) at 2 K/s in air (72.74 wt pct Fe2O3, 6.11 wt pct SiO2 and 21.14 wt pct CaO). (BSE)
L + C2S + CF2 + CF (Assemblage IV)
The final microstructures to form consisted of individual CF crystals (IVa), coupled C2S+CF microstructures (IVb) and a coupled C2S+CF2+CF microstructure (IVc). These phases are the last to form during or before the complete solidification of the liquid at between 1200 °C and 1195 °C.
The individual CF crystals (IVa) were observed to form interfaces with the individual CF2 crystals (IIIa), the coupled C2S-CF2 microstructure (IIIb), and the coupled C2S-CF (IVb) and coupled C2S-CF2-CF (IVc) microstructures. The interfaces between CF and CF2 were irregular and appeared as diffuse in SEM imaging. These individual crystals were smaller than the CF2 and C2S individual crystals present in the sample and constrained in all dimensions by the previously formed solids.
The fine interdispersion of the phases make it difficult to characterize individual phases in the coupled C2S-CF (IVb) and coupled C2S-CF2-CF (IVc) microstructures. The phases present in these two microstructures were determined by line analysis with EPMA. The diameter of the excitation volume created by the electron beam in these materials is of the order of 1 μm. As the position of the probe is moved across these fine microstructures, the mean composition therefore changes with the proportion of each material within the interaction volume. The measured compositions should however be on the binary join between the end members or within the alkemade triangles, assuming that the accuracy of the measurements is not affected by the heterogeneous nature of the material in the interaction volume. The line measurements were compared to the joins on the ternary phase diagram, as illustrated in Figure 11. Based on the line analysis, the four coupled microstructures were confirmed, C2S-Hematite (Microstructure IIc), C2S-CF2 (Microstructure IIIb), C2S-CF (Microstructure IVb), and C2S-CF2-CF (Microstructure IVc). The measured compositions for the C2S-CF2-CF coupled microstructure were found to be within the C2S-CF2-CF Alkemade triangle rather than along the C2S-CF or C2S-CF2 joins. This was attributed to either the presence of a ternary coupled microstructure or the interaction volume of the EPMA sampling two microstructures (e.g., both the C2S-CF2 and C2S-CF are sampled by the EPMA interaction volume) at the same time.

EPMA line analysis of coupled structures formed when cooling a liquid from 1623 K to 1073 K (1350 °C to 800 °C) at 2 K/s in air (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO)
The C2S-CF2-CF coupled microstructure and the C2S-CF coupled microstructures were both observed to share interfaces with the CF and CF2 individual crystals and the C2S-CF2 coupled microstructure. Both coupled microstructures were observed to consist of either rods of C2S (< 1 μm diameter) in a CF or CF2/CF matrix or as an irregular coupled microstructure. The C2S in this microstructure extended from surface microstructures to the coupled microstructure.
Liquid Composition
In the current study, the compositions of the liquid at selected temperatures were measured with EPMA. The measured liquid compositions are shown in Figure 12 and given in Table I.

Adapted from Ref. [19]
Measured liquid compositions for oxide samples of bulk composition X (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO, marked) cooled from 1623 K (1350 °C) at 2 K/s in comparison to the equilibrium liquidus for the “Fe2O3”-CaO-SiO2 system in air.
During the initial solidification of hematite (I), with decreasing temperature the liquid composition moved from the hematite primary phase field into the C2S primary phase field. As this occurred, some regions of the samples were observed to be of assemblage II (L + H + C2S). Once C2S was present, the coupled hematite + C2S microstructure (IIc) and individual C2S crystals (IId) solidified from the liquid. The liquid composition in Assemblage II (L + H + C2S) moved away from the C2S primary phase field as the temperature decreased. This continued as the liquid approached the CF2 primary phase field. In some regions of the sample, CF2 nucleated and these regions transitioned into Assemblage III (L + C2S + CF2). When compared to the composition of the liquid in the surrounding area (Assemblage II), only CF2 had solidified from the liquid in these regions (Assemblage III) (Table II).
The proportion of the phases in Assemblage II (L + H + C2S) at each temperature can be determined by undertaking a mass balance using the measured liquid composition, as summarized in Table 1. The calculated proportions of these phases present for this assemblage are shown in Figure 13. From this figure, it is seen that the proportion of hematite in the assemblage did not significantly change in the temperature range over which Assemblage II (L + H + C2S) was observed to form, only increased by 3 wt pct between 1508 K and 1498 K (1235 °C and 1225 °C). The C2S was observed to undergo a significant change, increasing from 0 to 10 wt pct between 1513 K and 1488 K (1240 °C and 1215 °C). The C2S increased from 0 to 8 wt pct between 1240 °C and 1225 °C. In this range, the increase in C2S with time was approximately constant, at approximately 1 wt pct/s. This could be taken to indicate that the C2S solidified independently from hematite from 1513 K and 1498 K (1240 °C to 1225 °C). At temperatures between 1498 K and 1488 K (1225 °C and 1215 °C), both the C2S and hematite solidified simultaneously.

Calculated proportions of phases with temperature for Assemblage II, as determined by mass balance with the measured liquid compositions
Discussion
There are significant differences between the observed microstructures and those predicted from both equilibrium and Scheil–Gulliver cooling. In the solidified material obtained by cooling the liquid from 1623 K to 1073 K (1350 °C to 800 °C) at 2 K/s;
-
1.
No SFC phase is formed at any stage
-
2.
The liquid composition on cooling does not follow that predicted by equilibrium or Sheil-Gulliver solidification
-
3.
There is no evidence of redissolution of solid phases once formed, as expected for equilibrium cooling and required for peritectic reactions
However, based on the experiments undertaken, the sequence of solidification and origins of microstructures can be determined. An example of the microstructures formed and the various phase assemblages contributing to it is illustrated in Figure 14.

Example of the microstructures observed in samples cooled from 1623 K to 1073 K (1350 °C to 800 °C) at 2 K/s in air (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO). (BSE)
Formation of Assemblage I (L + H)
Based on the results of the present study for the selected bulk composition and cooling rate, on initial cooling of the liquid, stoichiometric hematite is the first phase to be formed. Metallographic observations indicated that this takes place through heterogeneous nucleation on the platinum substrate and possibly later homogenously from the liquid. The proportion of hematite and the number of hematite crystals increases with decreasing temperature. The hematite typically forms skeletal faceted dendritic structures. Despite the fact that the liquid is subsequently cooled to below the SFC-Hematite peritectic temperature, continued solidification of the hematite takes place. The resulting liquid composition was found to move progressively from the hematite primary phase field through the SFC primary phase field and into the C2S primary phase field.
No SFC was observed within the sample when the liquid composition was cooling through the SFC primary phase field. It appears that it is more kinetically favorable to continue hematite solidification than to nucleate a new liquid/SFC interface.
The measured liquid compositions on cooling are illustrated in Figure 15, which represents the pseudo-ternary section of the “Fe2O3”-CaO-SiO2 system in air with constant CaO/SiO2 ratio of 3.46 wt/wt. At the highest Fe2O3 concentrations measured at each temperature, the liquid compositions appear to represent the metastable liquid in equilibrium with hematite in the absence of the equilibrium C2S phase. The experimental points included in the figure with arrows pointing to the right, the direction of higher Fe2O3 concentrations, represent samples /regions of the samples where there is evidence that some mass transfer of iron to the surfaces of existing hematite crystals has taken place during quenching. In these cases the Fe2O3 concentrations are lower than anticipated at the quench temperature, but have been included for completeness. A number of liquid compositions not believed to be representative of conditions at temperature have been presented in Figure 15, but not included in Table I.

Comparison of the measured liquid compositions for Assemblage I (L + H) with the non-equilibrium hematite (metastable) and C2S liquidus. (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO, air). This pseudo- binary section has a constant CaO/SiO2 ratio of 3.46 wt/wt
Since SFC is not formed on cooling, this primary phase field has been replaced in Figure 15 with the metastable hematite liquidus, which is close to but slightly above the SFC liquidus. It can be seen that the temperature at which the L + H and L + C2S liquidus isotherms intersects (approximately 1518 K, 1245 °C) differs from that at which the L + SFC and L + C2S liquidus isotherms intersect (approximately 1508 K, 1235 °C) in Figure 12. Consistent with the hematite liquidus, a decrease in temperature resulted in the removal of Fe2O3 from the liquid as a result of hematite formation, while maintaining a constant CaO/SiO2 ratio. The majority of the measured liquid compositions were lower in Fe2O3 than that predicted by equilibrium or the hematite liquidus extension. This was despite the measurements being performed in regions of the sample that appeared to be well quenched, that is close to the sample surface and far from solidified material. For hematite solidification to occur, the liquid composition should be richer in Fe2O3 than that indicated by the L+H liquidus isotherm at all temperatures. The fact that the measured compositions are lower in iron than anticipated indicates that some hematite continued to precipitate on quenching the samples.
During the final stages of Assemblage I, the liquid compositions were inside the C2S primary phase field, i.e., below the L+C2S liquidus, showing the supersaturation of the liquid with respect to C2S. Despite this, the hematite continued to solidify as cooling progressed.
Examining the shape and position of the metastable hematite liquidus extension it can be seen that it closely follows that of the equilibrium phase SFC liquidus. In effect the thermodynamic driving forces for both hematite and SFC formation from the melt, that is the Gibbs free energy difference between solid and liquid phases, are similar within this range of conditions, as evident by the similar liquidus temperatures. The crystal structure of the SFC phase is relatively complex compared to the orthorhombic hematite, which is close to a hexagonal close packed oxygen arrangement with interstitial ferric ions.[25] The SFC phase in contrast consists of alternating layers of spinel (M4T2O8) and pyroxene (M2Si4O12). This results in alternating layers of octahedral and tetrahedral layers, with the tetrahedral layer continuous in tetrahedral units and the octahedral layer consisting of ribbons 4 octahedral units wide.[26] It is anticipated that the nucleation frequency of this more complex crystal structure would be lower than that of hematite for the same bulk liquid composition for a given undercooling. This in part explains the absence of the SFC phase during cooling under the conditions investigated in the present study.
Formation of Assemblage II (L + H + C2S)
All phase assemblages and microstructures observed in Assemblage I and II are present in the total solidified material when Assemblages III and IV were present, indicating that all microstructures formed during this stage were a result of cooling, rather than quenching. As observed in Figure 12, the composition of the liquids in close proximity to Assemblage II was found to be within the C2S primary phase field. At the highest temperatures (1508 K to 1503 K, 1235 °C to 1230 °C), both Assemblage I and II were observed within a single sample. The two assemblages were observed to be isolated from each other and the liquid composition adjacent to these two separate assemblages was measured by EPMA. These compositions are given in Table I. At the highest temperatures in which Assemblage II is observed the liquid compositions were found to be close to those measured for Assemblage I. Repeat experiments confirmed that the appearance of Assemblage II at these temperatures was not an artifact of quenching. The proportion of Assemblage II relative to Assemblage I increased with decreasing temperature, until Assemblage I was no longer observed. At the highest temperatures in which Assemblage II is observed, the liquid composition was observed to be close to that measured for Assemblage I at the same temperature. As temperature decreased, the liquid compositions moved towards the C2S-SFC boundary curve. During the solidification of Assemblage II, both hematite and C2S were found to be solidifying, indicating that the liquid was supercooled with respect to both C2S and hematite. This is despite the liquid composition being in the C2S primary phase field on the ternary phase diagram, as illustrated by Figure 16. The exact composition/temperature trajectory from the undercooled melt in the C2S primary phase field to the peritectic temperature and the appearance of CF2 phase is not known at this time.

Schematic representation of the change in liquid composition as solidification occurred for Assemblages I and II assuming a metastable liquidus in which SFC is absent (72.74 wt pct Fe2O3, 6.11 wt pct SiO2 and 21.14 wt pct CaO)
Metallographic evidence indicates that a thin layer of C2S (IIa) nucleated heterogeneously and grew at the hematite-liquid interface or homogenously in liquid adjacent to the hematite-liquid interface. The other two microstructures, the individual C2S crystals (IIb) and the coupled C2S-H (IIc) microstructures, were subsequently formed.
From the calculated proportions of phases (Figure 13) and the liquid compositions (Table I) at each temperature, the order of formation of the remaining microstructures (IIb, IIc) can be determined, as illustrated by Figure 16. During the initial stage of solidification, the rate of C2S solidification was faster than hematite solidification, indicating that the coupled H+C2S microstructure (IIc) was not forming. At the highest temperatures, only the individual C2S crystals (IIb) were forming. As temperature decreased further, the rate C2S formation decreased and was similar to the rate of hematite formation. At the lower temperatures in which this assemblage was observed, the coupled C2S-hematite microstructure (IIc) was the predominant microstructure forming. The mass ratio of C2S/H in the coupled microstructure is approximately 0.7, which is consistent with the that expected from the phase diagram in Figure 12.
From the metallographic evidence it is concluded that C2S layer formed first (IIa), followed by the individual C2S crystals (IIb), and lastly the coupled H-C2S microstructure (IIc). The individual C2S crystals were not associated with hematite, indicating that the hematite was unable to nucleate on the C2S phase. The individual C2S crystals solidified as dendrites.
The microstructures formed in phase assemblage II can also be related to the kinetic phenomena taking place during cooling. It was demonstrated in Figure 15 that the undercooling of the liquid with respect to SFC is relatively small. Inspection of this figure shows that on continued cooling the undercooling with respect to the C2S phase increases as the temperature of the liquid decreases. For example, at 1493 K (1220 °C) and 32 wt pct CaO + SiO2, the undercooling with respect to the C2S liquidus is approximately 50 K, which is considerable. The nucleation and growth of the C2S from the liquid results in a the release of the heat of solidification The effect of the heat release counters the heat loss to the surrounds, locally changing the rate of cooling of the sample. The extent of this change is determined by the rates of heat conduction and convection within the liquid and solids, and the rate of solidification. This change in thermal conditions is illustrated schematically in Figure 17. The large thermodynamic driving force also leads to rapid C2S growth and the formation of the C2S crystal dendrite structure. As the C2S crystals are formed the local increase in temperature and the change in liquid composition lowers the thermodynamic driving force for C2S growth and the rate of growth of the individual crystals decrease. This then leads to the transition from single crystal C2S growth to the coupled growth of C2S+H and the formation of the binary eutectic structure.

The schematic representation of local temperature and time during C2S nucleation and growth Ref. [27]
Formation of Assemblage III (L + C2S + CF2)
The formation of assemblage III was found to take place over a small temperature range. The individual CF2 (IIIa) crystals were principally observed to be adjacent to hematite, suggesting heterogeneous nucleation of CF2 on the hematite. The CF2 was observed as individual CF2 crystals (IIIa) in all regions of assemblage III, while the coupled CF2-C2S microstructure (IIIb) was in limited areas. As such, this suggests that the CF2 individual crystals formed first in a liquid supercooled with respect to CF2, growing faster than the other phases present. These crystals were constrained by the microstructures formed in Assemblage I and II, solidifying around them. The shape of these crystals and the presence of splitting tips suggests that this phase solidified as a series of parallel plates rather than as individual crystal.
The coupled C2S-CF2 microstructure (IIIb) is believed to have formed after the individual CF2 crystals (IIIa), most likely via a coupled growth mechanism. This microstructure was observed to be associated with the individual CF2 crystals (IIIa), indicating that the C2S nucleated heterogeneously on the CF2 or in the liquid adjacent to the liquid-CF2 interface. Based on the microstructure and image analysis, it appears that there is larger proportion of CF2 than C2S in the coupled microstructure (see Figure 9). This is consistent with the ratio of the C2S/CF2, predicted to be 0.25 based on the phase diagram (Figure 12) and estimated to be 0.29 based on image analysis of the micrographs.
Based on the order of solidification, the liquid composition can be inferred followed a path similar to that illustrated in Figure 18. Prior to the formation of the CF2, within the CF2 primary phase field, the liquid composition would have been supersaturated with respect to CF2. Initially, the individual CF2 crystals (IIIa) are formed. At some point, C2S is nucleated on the CF2 Crystals (see Figure 18). Once the C2S-CF2 coupled microstructure (IIIb) started to form, the composition would have moved towards the C2S-CF2 boundary curve and the C2S-CF2-CF ternary eutectic point. These processes give two-step changes in the liquid composition. There are small differences and changes in the liquid compositions that could not be accurately measured in the present study primarily due to quenching difficulties and low volume fraction of liquid.

Schematic representation of the changes in liquid composition as solidification occurred for Assemblage III (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO)
Formation of Assemblage IV (C2S + CF2 + CF)
The CF was observed in the final microstructure, and present in three microstructures, individual CF crystals (IVa), a C2S-CF coupled microstructure (IVb), and a C2S-CF2-CF coupled microstructure (IVc). Based on the mechanisms observed for the other observed assemblages, the possible mechanism for the final stages of solidification can be inferred, as illustrated by Figure 19. The liquid composition during the three stages of solidification are illustrated schematically in Figure 19. The order of formation is likely to be individual CF crystals (IVa), the coupled C2S-CF microstructure and finally the coupled C2S-CF2-CF microstructure. Its likely that the CF formed from a liquid supercooled with respect to CF, with solidification initially forming individual crystals of CF (IVa). Once the individual CF crystals solidified, the liquid composition would have decreased in CaO and Fe2O3 in equal proportions on a molar basis. The CF phase appears to have nucleated heterogeneously on CF-2. The remaining liquid would have solidified to form the C2S-CF coupled microstructure (IVb) via a coupled growth mechanism and finally the C2S-CF2-CF coupled microstructure (IVc) with the same mechanism. Once the formation of the C2S-CF microstructure was initiated, the liquid composition would have moved towards the C2S-CF boundary curve and towards the C2S-CF2-CF ternary eutectic point, where the final solidification (IVb) would have occurred (Figure 19).

The change in liquid composition as solidification occurred for Assemblage IV (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO)
Overall Impacts of Kinetics
The results of the present study have demonstrated that kinetic factors can play a significant role in determining the phases and microstructures formed on the solidification of these oxide liquids. Whilst most of the reactions and resultant product morphologies are consistent with those anticipated from solidification theory, for example, the formation of dendritic structures, pseudo-binary and ternary eutectics, the outstanding feature not previously anticipated was the role of nucleation (Figure 20).

The cumulative phase assemblages and microstructures during the formation of the four phase assemblages (72.74 wt pct Fe2O3, 6.11 wt pct SiO2, and 21.14 wt pct CaO), formed from the liquid on cooling in air at 2 K/s
In the system, and for the bulk composition selected for study, it was anticipated from the equilibria studies that the SFC phase would be formed on cooling of the liquids, however this was not the case. Nowhere in the solidification sequence was this phase formed. In addition, considerable undercoolings of the liquids were observed before the formation of the dicalcium silicate, calcium diferrite and calcium ferrite phases. These phenomena resulted in the formation of complex metastable phase assemblages. The rates of diffusional mass transfer of species in these low-silica ionic liquids are high relative to high-silica liquids.[28] The formation of these non-equilibrium structures is then all the more unexpected.
In the present study, only a single cooling rate was investigated. The results, however, indicate that the thermal history is a potentially important factor influencing the resultant sinter microstructures and phase assemblages.
Summary
A new experimental technique has been developed that enables the cooling rates of oxide liquids to be accurately controlled. Using this new approach, the sequence of phase formation and the product microstructures formed in specific temperature ranges can be identified.
The solidification of liquids in the system “Fe2O3”-CaO-SiO2 having a bulk composition with a CaO/SiO2 ratio of 3.46 and 73.17 wt pct Fe2O3, within the hematite primary phase field and predicted to form SFC during equilibrium have been studied. The experiments were carried out in air and with a cooling rate of 2 K/s. The phases present in the final microstructure were, hematite, C2S, CF and CF2. No SFC was formed indicating non-equilibrium cooling. The solidification sequence was determined to be as follows: nucleation and growth of the primary phase hematite, nucleation of C2S followed by the formation of a C2S layer on hematite, C2S dendrites and a coupled C2S-hematite microstructure, nucleation of CF2 followed by the formation of individual CF2 crystals and a coupled CF2-C2S microstructure, and finally the nucleation of CF followed by the formation of individual CF crystals and coupled C2S-CF and C2S-CF-CF2 microstructures.
The study clearly demonstrates the importance of thermal history in determining phase formation and product microstructure during solidification of these ‘Fe2O3”-CaO-SiO2 liquids. This observation is significant in that it provides valuable insight into the key processes taking place during industrial iron ore sintering.
References
L. Lu and O. Ishiyama: in Iron Ore, Elsevier Ltd, 2015, pp. 395–433.
R. Lloyd, Min. Metall. 1922, vol. 3 (190), pp. 17–19.
J. Ostwald, BHP Tech. Bull. 1981, vol. 25, pp. 13-20.
C. Loo, K. Wan and V. Howes, Ironmaking Steelmaking 1988, vol. 15, pp. 279-285.
D. Fernández-González, I. Ruiz-Bustinza, J. Mochón, C. González-Gasca and L. Verdeja, Miner. Process. Extr. Metall. Rev. 2017, vol. 38, pp. 254-264.
C. Loo and W. Leung, ISIJ Int. 2003, vol. 43, pp. 1393-1402.
L. Yang and L. Davis, ISIJ Int. 1999, vol. 39, pp. 239-245.
P Dawson, J Ostwald and K Hayes, Trans. Inst. Min. Metall., Sect. C 1985, vol. 94, pp. C71-C78.
R. Burlingame, G. Bitsianes and T. Joseph, J. Met. 1956, vol. 8 (7), pp. 853-861.
I. Shigaki, M. Sawada, O. Tsuchiya, K. Yoshioka and T. Takahashi, Tetsu to Hagane 1984, vol. 70, pp. 2208-2215.
Y. Hida, J. Okazaki, K. Otoh and M. Sasaki, Tetsu to Hagane 1987, vol. 73, pp. 1893-1900.
E. Marliere, J. Steiler and D. Jeulin, in Proceedings of the Process Technology Conference, 1986, pp 523–35.
N. Scarlett, I. Madsen, M. Pownceby and A. Christensen, J. Appl. Crystallogr. 2004, vol. 37, pp. 362-368.
N. Scarlett, M. Pownceby, I. Madsen and A. N. Christensen, Metall. Mater. Trans. B 2004, vol. 35B, pp. 929–936.
N. Webster, M. Pownceby, I. Madsen and J. Kimpton, Metall. Mat. Trans. B 2012, vol. 43, pp. 1344-1357.
N. Webster, M. Pownceby and I. Madsen, ISIJ Int. 2013, vol. 53, pp. 1334-1340.
N. Webster, M. Pownceby, I. Madsen and J. Kimpton, ISIJ Int. 2013, vol. 53, pp. 774-781.
M. Pownceby, N. Webster, J. Manuel and N. Ware, Trans. Inst. Min. Metall. Sect. C 2016, vol. 125, pp. 140–148.
J. Chen, M. Schevchenko, P. Hayes and E. Jak, ISIJ Int. 2019, vol. 59, pp. 795-804.
B. Phillips and A. Muan, J. Am. Ceram. Soc. 1959, vol. 42, pp. 413-23.
M. Pownceby, J. Clout and M. Fisher-White, Trans. Inst. Min. Metall. Sect. C 1998, vol. 107, pp. C1-C10.
S. Nicol, E. Jak, J. Chen, W. Qi, X. Mao and P. Hayes, in 6th Baosteel Australian Joint Centre Conf., Wollongong, 2018.
D. Ball: Agglomeration of iron ores. London : Heinemann Educational, London, 1973.
F. Incropera: Fundamentals of heat and mass transfer. 6th ed.. ed. Hoboken, NJ : John Wiley, Hoboken, NJ, 2007.
R. Blake, R. Hessevick, T. Zoltai and L. Finger, Am. Mineral. 1966, vol. 51, pp. 123-129.
J. Hamilton, B. Hoskins, W. Mumme, W. Borbidge and M. Montague, Neues Jahrb. Mineral. Abh. 1989, vol. 161, pp. 1-26.
H. Fredriksson and U. Åkerlind: Solidification and Crystallization Processing in Metals and Alloys. Chichester, UK: John Wiley & Sons, Ltd, Chichester, UK, 2012.
Y. Waseda and J. Toguri: The Structure and Properties of Oxide Melts : Application of Basic Science to Metallurgical Processing. Singapore River Edge, NJ : World Scientific, 1998.
Acknowledgments
The authors would like to thank BHP Billiton for financial support to enable this research to be carried out, and the Centre of Microstructure and Microanalysis (CMM), the University of Queensland for providing electron microscope facilities that enabled the microanalytical measurements to be undertaken. This research was supported by an Education Endowment Fund (EEF) scholarship from the Australasian Institute of Mining and Metallurgy (AusIMM) and an Australian Government Research Training Program (RTP) Scholarship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted March 27, 2019.
Rights and permissions
About this article

Cite this article
Nicol, S., Jak, E. & Hayes, P.C. Microstructure Evolution During Controlled Solidification of “Fe2O3”-CaO-SiO2 Liquids in Air. Metall Mater Trans B 50, 2706–2722 (2019). https://doi.org/10.1007/s11663-019-01687-w
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-019-01687-w