
Abstract
Ciliopathy encompasses a diverse group of autosomal recessive genetic disorders caused by mutations in genes coding for components of the primary cilia. Skeletal ciliopathy forms a subset of ciliopathies characterized by distinctive skeletal changes. Common skeletal ciliopathies include Jeune asphyxiating thoracic dysplasia, Ellis–van Creveld syndrome, Sensenbrenner syndrome, and short-rib polydactyly syndromes. These disorders share common clinical and radiological features. The clinical hallmarks comprise thoracic hypoplasia with respiratory failure, body disproportion with a normal trunk length and short limbs, and severely short digits occasionally accompanied by polydactyly. Reflecting the clinical features, the radiological hallmarks consist of a narrow thorax caused by extremely short ribs, normal or only mildly affected spine, shortening of the tubular bones, and severe brachydactyly with or without polydactyly. Other radiological clues include trident ilia/pelvis and cone-shaped epiphysis. Skeletal ciliopathies are commonly associated with extraskeletal anomalies, such as progressive renal degeneration, liver disease, retinopathy, cardiac anomalies, and cerebellar abnormalities. In this article, we discuss the radiological pattern recognition approach to skeletal ciliopathies. We also describe the clinical and genetic features of skeletal ciliopathies that the radiologists should know for them to play an appropriate role in multidisciplinary care and scientific advancement of these complicated disorders.














Similar content being viewed by others
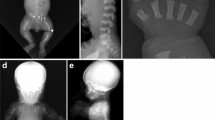
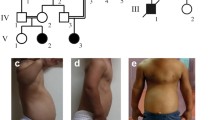
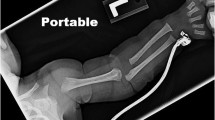
References
Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet. 2012;160(3):165–74.
Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum Mutat. 2018;39(1):152–66.
Yuan X, Serra RA, Yang S. Function and regulation of primary cilia and intraflagellar transport proteins in the skeleton. Ann N Y Acad Sci. 2015;1335:78–99.
Schmidts M. Clinical genetics and pathobiology of ciliary chondrodysplasias. J Pediatric Genet. 2014;3(2):46–944.
Hammarsjo A, Wang Z, Vaz R, Taylan F, Sedghi M, Girisha KM, et al. Novel KIAA0753 mutations extend the phenotype of skeletal ciliopathies. Sci Rep. 2017;7(1):15585.
Dagoneau N, Goulet M, Genevieve D, Sznajer Y, Martinovic J, Smithson S, et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am J Hum Genet. 2009;84(5):706–11.
Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167(12):2869–92.
Spranger J. Pattern recognition in bone dysplasias. Prog Clin Biol Res. 1985;200:315–42.
Spranger J. Bone dysplasia 'families'. Pathol Immunopathol Res. 1988;7(1–2):76–80.
Jeune M, Beraud C, Carron R. Asphyxiating thoracic dystrophy with familial characteristics. Arch Fr Pediatr. 1955;12(8):886–91.
Oberklaid F, Danks DM, Mayne V, Campbell P. Asphyxiating thoracic dysplasia Clinical, radiological, and pathological information on 10 patients. Arch Dis Child. 1977;52(10):758–65.
Keppler-Noreuil KM, Adam MP, Welch J, Muilenburg A, Willing MC. Clinical insights gained from eight new cases and review of reported cases with Jeune syndrome (asphyxiating thoracic dystrophy). Am J Med Genet A. 2011;155(5):1021–32.
Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011;26(7):1039–56.
Aurora P, Wallis CE. Jeune syndrome (asphyxiating thoracic dystrophy) associated with Hirschsprung disease. Clin Dysmorphol. 1999;8(4):259–63.
Tuz K, Bachmann-Gagescu R, O'Day DR, Hua K, Isabella CR, Phelps IG, et al. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am J Hum Genet. 2014;94(1):62–72.
Shaheen R, Shamseldin HE, Loucks CM, Seidahmed MZ, Ansari S, Ibrahim Khalil M, et al. Mutations in CSPP1, encoding a core centrosomal protein, cause a range of ciliopathy phenotypes in humans. Am J Hum Genet. 2014;94(1):73–9.
Pirnar T, Neuhauser EB. Asphyxiating thoracic dystrophy of the newborn. Am J Roentgenol Radium Ther Nucl Med. 1966;98(2):358–64.
Leonard O, Langer J. Thoracic–pelvic–phalangeal dystrophy. Radiology. 1968;91(3):447–56.
Shaheen R, Schmidts M, Faqeih E, Hashem A, Lausch E, Holder I, et al. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum Mol Genet. 2015;24(5):1410–9.
Cavalcanti DP, Huber C, Sang KH, Baujat G, Collins F, Delezoide AL, et al. Mutation in IFT80 in a fetus with the phenotype of Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff dysplasia spectrum. J Med Genet. 2011;48(2):88–92.
Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013;93(5):915–25.
Ellis RW, van Creveld S. A syndrome characterized by ectodermal dysplasia, polydactyly, chondro-dysplasia and congenital morbus cordis: report of three cases. Arch Dis Child. 1940;15(82):65–84.
Weller SD. Chondro-ectodermal dysplasia (Ellis-Van Creveld syndrome). Proc R Soc Med. 1951;44(8):731–2.
McKusick VA, Egeland JA, Eldridge R, Krusen DE. Dwarfism in the amish I. The Ellis-Van creveld syndrome. Bull Johns Hopkins Hosp. 1964;115:306–36.
da Silva EO, Janovitz D, de Albuquerque SC. Ellis-van creveld syndrome: report of 15 cases in an inbred kindred. J Med Genet. 1980;17(5):349–56.
Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, et al. Mutations in a new gene in Ellis-van creveld syndrome and Weyers acrodental dysostosis. Nat Genet. 2000;24(3):283–6.
Galdzicka M, Patnala S, Hirshman MG, Cai JF, Nitowsky H, Egeland JA, et al. A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Mol Genet Metab. 2002;77(4):291–5.
Tompson SW, Ruiz-Perez VL, Blair HJ, Barton S, Navarro V, Robson JL, et al. Sequencing EVC and EVC2 identifies mutations in two-thirds of Ellis-van Creveld syndrome patients. Hum Genet. 2007;120(5):663–70.
Caparros-Martin JA, De Luca A, Cartault F, Aglan M, Temtamy S, Otaify GA, et al. Specific variants in WDR35 cause a distinctive form of Ellis-van Creveld syndrome by disrupting the recruitment of the EvC complex and SMO into the cilium. Hum Mol Genet. 2015;24(14):4126–37.
Sensenbrenner JA, Dorst JP, Owens RP. New syndrome of skeletal, dental and hair anomalies. Birth Defects Orig Artic Ser. 1975;11(2):372–9.
Levin LS, Perrin JC, Ose L, Dorst JP, Miller JD, McKusick VA. A heritable syndrome of craniosynostosis, short thin hair, dental abnormalities, and short limbs: cranioectodermal dysplasia. J Pediatr. 1977;90(1):55–61.
Zaffanello M, Diomedi-Camassei F, Melzi ML, Torre G, Callea F, Emma F. Sensenbrenner syndrome: a new member of the hepatorenal fibrocystic family. Am J Med Genet A. 2006;140(21):2336–400.
Amar MJ, Sutphen R, Kousseff BG. Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome). Am J Med Genet. 1997;70(4):349–52.
Bacino CA, Dhar SU, Brunetti-Pierri N, Lee B, Bonnen PE. WDR35 mutation in siblings with Sensenbrenner syndrome: a ciliopathy with variable phenotype. Am J Med Genet A. 2012;158(11):2917–24.
Miyazaki O, Nishimura G, Kagami M, Ogata T. Radiological evaluation of dysmorphic thorax of paternal uniparental disomy 14. Pediatr Radiol. 2011;41(8):1013–9.
Moosa S, Obregon MG, Altmuller J, Thiele H, Nurnberg P, Fano V, et al. Novel IFT122 mutations in three Argentinian patients with cranioectodermal dysplasia: expanding the mutational spectrum. Am J Med Genet A. 2016;170(5):1295–301.
Silveira KC, Moreno CA, Cavalcanti DP. Beemer-Langer syndrome is a ciliopathy due to biallelic mutations in IFT122. Am J Med Genet A. 2017;173(5):1186–9.
Saldino RM, Noonan CD. Severe thoracic dystrophy with striking micromelia, abnormal osseous development, including the spine, and multiple visceral anomalies. Am J Roentgenol Radium Ther Nucl Med. 1972;114(2):257–63.
Verma IC, Bhargava S, Agarwal S. An autosomal recessive form of lethal chondrodystrophy with severe thoracic narrowing, rhizoacromelic type of micromelia, polydacytly and genital anomalies. Birth Defects Orig Artic Ser. 1975;11(6):167–74.
Naumoff P, Young LW, Mazer J, Amortegui AJ. Short rib-polydactyly syndrome type 3. Radiology. 1977;122(2):443–7.
Huber C, Wu S, Kim AS, Sigaudy S, Sarukhanov A, Serre V, et al. WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-kappaB pathway in cilia. Am J Hum Genet. 2013;93(5):926–31.
McInerney-Leo AM, Schmidts M, Cortes CR, Leo PJ, Gener B, Courtney AD, et al. Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am J Hum Genet. 2013;93(3):515–23.
Spranger JW, Hall C, Nishimura G, Superti-Furga A, Unger S. Bone dysplasias. 3rd ed. New York: Oxford University Press; 2012.
Majewski F, Pfeiffer RA, Lenz W, Müller R, Feil G, Seiler R. Polysyndaktylie, verkürzte Gliedmaßen und Genitalfehlbildungen: Kennzeichen eines selbständigen Syndroms? Z Kinderheilkd. 1971;111(2):118–38.
Baraitser M, Burn J, Fixsen J. A female infant with features of Mohr and Majewski syndromes: variable expression, a genetic compound, or a distinct entity? J Med Genet. 1983;20(1):65–7.
Burn J, Dezateux C, Hall CM, Baraitser M. Orofaciodigital syndrome with mesomelic limb shortening. J Med Genet. 1984;21(3):189–92.
El Hokayem J, Huber C, Couve A, Aziza J, Baujat G, Bouvier R, et al. NEK1 and DYNC2H1 are both involved in short rib polydactyly Majewski type but not in Beemer Langer cases. J Med Genet. 2012;49(4):227–33.
Thomas S, Legendre M, Saunier S, Bessieres B, Alby C, Bonniere M, et al. TCTN3 mutations cause Mohr–Majewski syndrome. Am J Hum Genet. 2012;91(2):372–8.
Beemer FA, Langer LO Jr, Klep-de Pater JM, Hemmes AM, Bylsma JB, Pauli RM, et al. A new short rib syndrome: report of two cases. Am J Med Genet. 1983;14(1):115–23.
Bizaoui V, Huber C, Kohaut E, Roume J, Bonniere M, Attie-Bitach T, et al. Mutations in IFT80 cause SRPS type IV Report of two families and review. Am J Med Genet A. 2019;179(4):639–44.
Mainzer F, Saldino RM, Ozonoff MB, Minagi H. Familial nephropathy associated with retinitis pigmentosa, cerebellar ataxia and skeletal abnormalities. Am J Med. 1970;49(4):556–62.
Giedion A. Phalangeal cone shaped epiphysis of the hands (PhCSEH) and chronic renal disease—the conorenal syndromes. Pediatr Radiol. 1979;8(1):32–8.
Ehara S, Kim OH, Maisawa S, Takasago Y, Nishimura G. Axial spondylometaphyseal dysplasia. Eur J Pediatr. 1997;156(8):627–30.
Wang Z, Horemuzova E, Iida A, Guo L, Liu Y, Matsumoto N, et al. Axial spondylometaphyseal dysplasia is also caused by NEK1 mutations. J Hum Genet. 2017;62(4):503–6.
Oud MM, Lamers IJ, Arts HH. Ciliopathies: genetics in pediatric medicine. J Pediatr Genet. 2017;6(1):18–29.
Ehlen HW, Buelens LA, Vortkamp A. Hedgehog signaling in skeletal development. Birth Defects Res Part C Embryo Today Rev. 2006;78(3):267–79.
Kunova Bosakova M, Varecha M, Hampl M, Duran I, Nita A, Buchtova M, et al. Regulation of ciliary function by fibroblast growth factor signaling identifies FGFR3-related disorders achondroplasia and thanatophoric dysplasia as ciliopathies. Hum Mol Genet. 2018;27(6):1093–105.
Martin L, Kaci N, Estibals V, Goudin N, Garfa-Traore M, Benoist-Lasselin C, et al. Constitutively-active FGFR3 disrupts primary cilium length and IFT20 trafficking in various chondrocyte models of achondroplasia. Hum Mol Genet. 2018;27(1):1–13.
Wang C, Yuan X, Yang S. IFT80 is essential for chondrocyte differentiation by regulating Hedgehog and Wnt signaling pathways. Exp Cell Res. 2013;319(5):623–32.
Acknowledgements
Part of this article was originally presented at the 104th annual meeting of the Radiological Society of North America (RSNA) in Chicago, 2018. It received a Certification of Merit for an education exhibit.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Handa, A., Voss, U., Hammarsjö, A. et al. Skeletal ciliopathies: a pattern recognition approach. Jpn J Radiol 38, 193–206 (2020). https://doi.org/10.1007/s11604-020-00920-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11604-020-00920-w