
Abstract
Ceramicines are a series of limonoids which were isolated from the bark of Malaysian Chisocheton ceramicus (Meliaceae) and show various biological activities. Ceramicine B, in particular, has been reported to show a strong lipid droplet accumulation (LDA) inhibitory activity on a mouse pre-adipocyte cell line (MC3T3-G2/PA6). With the purpose of discovering compounds with stronger activity than ceramicine B, we further investigated the constituents of C. ceramicus. As a result, from the bark of C. ceramicus four new ceramicines (ceramicines M–P, 1–4) were isolated, and their structures were determined on the basis of NMR and mass spectroscopic analyses in combination with NMR chemical shift calculations. LDA inhibitory activity of 1–4 was evaluated. Compounds 1–3 showed LDA inhibitory activity, and 3 showed better selectivity than ceramicine B while showing activity at the same order of magnitude as ceramicine B. Since 3, which possess a carbonyl group at C-7, showed better selectivity than 5, which possess a 7α-OH group, while showing activity at the same order of magnitude as 5, we also investigated the effect of the substituent at C-7 by synthesizing several derivatives and evaluating their LDA inhibitory activity. Accordingly, we confirmed the importance of the presence of a 7α-OH group to the LDA inhibitory activity.
Similar content being viewed by others
Introduction
Ceramicines are a series of limonoids which were isolated in our previous phytochemical study on the bark of Malaysian Chisocheton ceramicus (Meliaceae). To date, 12 compounds were known, ceramicines A–L [1,2,3,4]. Ceramicines have been reported to show cytotoxic activity against a murine leukemia cell line (P388), potent anti-plasmodial activity against Plasmodium falciparum 3D7, lipid droplet accumulation (LDA) inhibitory activity on a mouse pre-adipocyte cell line (MC3T3-G2/PA6), and anti-melanin deposition activity against B16-F10 melanoma cells [1, 2, 5,6,7].
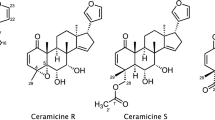
With the purpose of discovering compounds with stronger LDA inhibitory activity than ceramicine B (5), we further investigated the constituents of C. ceramicus. As a result, four new ceramicines (ceramicines M–P, 1–4, Fig. 1) were isolated and their structures were determined on the basis of NMR and mass spectroscopic analyses in combination with NMR chemical shift calculations. In addition, on the basis of the LDA inhibitory activity of the isolated compounds, we further investigated the effect of the substituent at C-7 of 5.

Structures of 1–5
Results and discussions
Structure elucidation of ceramicines M–P (1–4)
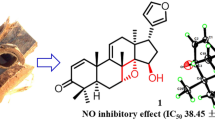
Ceramicine M (1) was obtained as an optically active, [α] 30D −47 (c 1.0, CHCl3), white amorphous solid and was revealed to have the molecular formula C24H28O5 by HRESITOFMS [m/z 419.1823 (M + Na)+, Δ −1.1 mmu]. IR absorptions implied the presence of α,β-unsaturated ketone (1690 cm−1) and hydroxy (3350 cm−1) groups. 1H and 13C NMR data (Table 1) revealed 24 carbon resonances due to one carbonyl, two sp 2 quaternary carbons, four sp 3 quaternary carbons, six sp 2 methines, five sp 3 methines, three sp 3 methylenes, and three methyls. Among them, three sp 3 carbons (δ C 73.5, 92.4, and 101.7) and two sp 2 methines (δ C 141.1 and 143.8) were ascribed to those bearing an oxygen atom.
Analyses of the HSQC and 1H–1H COSY spectra (Fig. 2) revealed the presence of four partial structures: a (C-2 and C-3), b (C-9, C-11, and C-12), c (C-15 to C-17), and d (C-22 and C-23). HMBC correlations of H3-18 to C-12, C-13, C-14, and C-17 suggested the connectivity of b, c, C-14, and C-18 through C-13. HMBC correlations of H-17 to C-20, C-21, and C-22, and H-23 to C-20 and C-21 suggested the presence of β-furyl at C-17, and the correlation of H2-16 to C-14 completed the structure of ring D. The presence of ring C was deduced from the HMBC cross-peaks of H3-30 to C-7, C-8, C-9, and C-14, and the connectivity of b, C-1, C-5, and C-19 through C-10 was suggested by the HMBC correlations of H3-19 to C-1, C-5, C-9, and C-10. HMBC correlations of H-2 to C-10 and C-4, and H-3 to C-1 and C-5 suggested the presence of ring A. Finally, HMBC correlations of H-6 to C-4, C-5, and C-7, and H-7 to C-4, and the chemical shift of C-4 (δ C 101.7) suggested the planar structure of 1 to be as shown in Fig. 2.

Selected 2D NMR correlations of 1
The relative configuration of 1 was assigned by analyses of the 1H–1H coupling constant data and the NOESY correlations (Fig. 3). First, H-6, H-17, CH3-19, and CH3-30 were assigned to be β-axially oriented from the NOESY correlations of H-6/H3-19 and H3-30, and H-12a/H-17 and H3-30, while H-9 and CH3-18 were deduced to possess α-orientation from the NOESY correlations of H3-18/H-9 and H-12b. Both H-5 and H-7 should possess β-orientation since C-4 and C-7 can only be connected through an ether linkage, and the multiplicity pattern of H-6 (br s) further supports this assumption.

Selected NOESY correlations of 1
Ceramicine N (2) was obtained as an optically active, [α] 31D +50 (c 1.0, CHCl3), white amorphous solid and was revealed to have the molecular formula C26H32O5 by HRESITOFMS [m/z 447.2157 (M + Na)+, Δ +1.0 mmu]. IR absorptions implied the presence of α,β-unsaturated ketone (1680 cm−1) and hydroxy (3480 cm−1) groups. 1H and 13C NMR data (Table 1) of 2 were highly similar to those of 5 [2]. In comparison to 5, the 1H and 13C NMR data of 2 showed an oxymethine signal (δ H 3.70, δ C 63.9) and sp 3 quaternary carbon (δ C 80.4) in place of a double bond signals (δ H 5.59, δ C 120.4, and δ C 159.8), and shifts of 1H and 13C signals of ring D. These data suggested 2 to be the 14,15-epoxy derivative of 5. The presence of the 14,15-epoxy moiety in 2 was also confirmed by HMBC correlations of H2-16 and H3-30 to δ C 80.4 (C-14) and 1H–1H COSY correlation of H-16b with δ H 3.70 (H-15). The orientation of the epoxy group was assumed to be α on the basis of the NOESY correlation of H-15/H3-30 and the multiplicity of H-15 (d, 2.8 Hz). In the case of a β-oriented epoxy group, the multiplicity of H-15 would be a triplet. This assumption was further supported by DFT NMR chemical shift calculations of the two possible isomers, 2a with α-oriented epoxy group and 2b with β-oriented epoxy group. As can be seen in Table 2, among the two possible isomers 2a gave the smallest mean average difference (MAD) and root-mean-square difference (RMSD) between the calculated and experimental chemical shifts, indicating the 2a as the more likely structure.
Ceramicine O (3) was obtained as an optically active, [α] 30D +10 (c 1.0, CHCl3), white amorphous solid and was revealed to have the molecular formula C26H30O4 by HRESITOFMS [m/z 429.2045 (M + Na)+, Δ +0.3 mmu]. IR absorptions implied the presence of ketone (1730 and 1680 cm−1) and hydroxy (3480 cm−1) groups. 1H and 13C NMR data (Table 1) of 3 were highly similar to those of 5 [2]. In comparison to 5, the 1H and 13C NMR data of 3 showed a carbonyl signal (δ C 205.6) in place of an oxymethine (δ H 4.23, δ C 72.5), and downfield shifts of CH-6 and C-8 signals. These data suggested 3 to be the 7-oxo derivative of 5. The presence of a carbonyl at C-7 in 3 was also confirmed by HMBC correlations of H-5 and H3-30 to δ C 205.6 (C-7). The relative configurations of 3 were deduced to be similar to those of 5 on the basis of the 1H–1H coupling constant data and NOESY correlations.
Ceramicine P (4) was obtained as an optically active, [α] 31D +34 (c 1.0, CHCl3), white amorphous solid and was revealed to have the molecular formula C27H32O6 by HRESITOFMS [m/z 475.2111 (M + Na)+, Δ +1.4 mmu]. IR absorptions implied the presence of ketones (1740 and 1690 cm−1) and hydroxy (3480 cm−1) groups. 1H and 13C NMR data (Table 1) of 4 were highly similar to those of ceramicine E [3]. In contrast to ceramicine E, the 1H and 13C NMR data of 4 showed two olefinic methine signals (δ H 6.01, δ C 130.9 and δ H 6.24, δ C 146.8) instead of two oxymethine signals (δ H 3.01, δ C 57.5 and δ H 3.35, δ C 52.9), indicating the presence of an α,β-unsaturated ketone in 4 in place of an α,β-epoxy ketone in ceramicine E. In addition, the chemical shifts of H-6 (δ H 4.18) and H-7 (δ H 5.38) of 4 indicate the presence of an acetyl at C-7. The structure of 4 was further confirmed by analyses of its 2D NMR data. In particular, HMBC correlations of H2-29 to δ C 146.8 (C-3), H3-30 with δ C 75.0 (C-7), and H-7 to δ C 170.8 (COMe) confirmed the presence of an α,β-unsaturated ketone and the acetyl position. The relative configurations of 4 were deduced to be similar to those of ceramicine E on the basis of the 1H–1H coupling constant data and NOESY correlations.
The absolute configurations of the isolated compounds were assumed to be similar to those of the previously reported ceramicines. The similarities of the CD spectra of 2 and 3 with those of ceramicine B, and 4 with ceramicine E further support this assumption.
The isolated compounds were tested for LDA inhibitory activity on MC3T3-G2/PA6 cells. As can be seen in Table 3, 1, 2, and 4 are less potent than 5. However, 3 showed better selectivity than 5 while showing activity at the same order of magnitude as 5.
Syntheses of ceramicine B derivatives
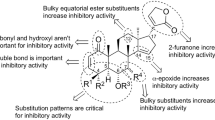
We have previously reported that the etherification or esterification of the 7α-OH group resulted in the increase of the cytotoxicity or decrease of LDA inhibitory activity [5]. In this work, we found that 3 with a carbonyl moiety at C-7 showed better selectivity than 5 while showing activity at the same order of magnitude as 5. Thus, we decided to further investigate the effect of the substituent at C-7 of 5.
First, we synthesized 7-dehydroxyceramicine B (6) and 7-epi-ceramicine B (7). Compound 6 was readily obtained through Barton–McCombie deoxygenation of 5 (Scheme 1) [8]. Since inversion of the configuration at C-7 could not be achieved through Mitsunobu reaction, we examined the feasibility of obtaining 7 through reduction of compound 3, which can be easily obtained from 5 after oxidation pyridinium chlorochromate (PCC). We used the Meerwein–Ponndorf–Verley (MPV) reduction to selectively reduce the carbonyl at C-7 [9], and as can be seen in Table 4, we obtained 7, albeit in a low yield. Interestingly, a ring-contracted side product 8 can also obtained using the MPV reduction (Table 4 entry 2). After obtaining 7, we synthesized its derivatives 9–11 (Scheme 2) to study the effects of etherification and esterification of the 7β-OH on the LDA inhibitory activity.

Synthesis of 6


Synthesis of 9–11. Reagents and conditions: a Ac2O, pyridine, DMAP, rt, 15 min, 64%; b BzCl, pyridine, DMAP, rt, 2 h, 32%; c EtI, NaH, DMF, rt, 4 h, 75%
The LDA inhibitory activities of the synthesized compounds are shown in Table 3. Compounds 6 and 7 have lower LDA inhibitory and cytotoxic activities than 5. Thus, the absence of an α-oriented hydroxy group at C-7 led to a decrease of both the LDA inhibitory and cytotoxic activities.
The effects of esterification and etherification of 7-OH group on the LDA inhibitory activity are as follow. On the basis of the IC50 values of 7 and 9–11, for the 7β-OH derivatives, esterification and etherification did not significantly change the LDA inhibitory activities. In contrast, on the basis of the IC50 values of 5 and 12–14, for the 7α-OH derivatives, esterification and etherification led to lower LDA inhibitory activities.
In addition, compound 8, with a contracted B-ring, showed no LDA inhibitory activity at 50 µM.
Experimental section
General experimental procedures
Optical rotations were measured on a JASCO DIP-1000 polarimeter. UV spectra were recorded on a Shimadzu UVmini-1240 spectrophotometer and IR spectra on a JASCO FT/IR-4100 spectrophotometer. High-resolution ESI MS were obtained on a LTQ Orbitrap XL (Thermo Scientific). 1H and 2D NMR spectra were measured on a 400-MHz or 600-MHz spectrometer at 300 K, while 13C NMR spectra were measured on a 100-MHz or 150-MHz spectrometer. The residual solvent peaks were used as internal standards (δ H 7.26 and δ C 77.0 for CDCl3, δ H 3.31 and δ C 49.0 for CD3OD). Standard pulse sequences were used for the 2D NMR experiments. Merck silica gel 60 (40–63 µm) was used for the column chromatography, and the separations were monitored by Merck silica gel 60 F254, or Merck silica gel RP C-18 F254 TLC plates.
Material
The bark of C. ceramicus was collected in Terengganu, Malaysia in July 2013. The botanical identification was made by Prof. A. Hamid A. Hadi, University of Malaya. Voucher specimens (No. HOSHI13CCB) are deposited in the department of pharmacognosy Hoshi University.
Extraction and isolation
The bark of C. ceramicus (8 kg) was extracted with MeOH to obtain 1.43 kg of extract. The MeOH extract was successively partitioned with n-hexane, EtOAc, n-BuOH, and water. The n-hexane-soluble materials were further separated by silica gel column chromatography (n-hexane/EtOAc 1:0 → 1:1, CHCl3/MeOH 1:0 → 0:1) to obtain 10 fractions (A–J). Fraction I was further separated with an ODS silica gel column (MeOH/H2O 7:3 → 1:0, acetone) to obtain six fractions (I-1 to I-6). Fraction I-2 (795 mg) was then separated by HPLC (Shiseido ODS MGII 30 × 250 mm, 75% MeOH(aq) at 8.0 mL/min, UV detection at 210 nm) into nine fractions (I-2-a to I-2-i). Separation of fraction I-2-d by HPLC (Shiseido ODS MGII 4.6 × 250 mm, 50% MeCN(aq) at 0.5 mL/min, UV detection at 210 nm) yielded 1 (1.1 mg, 0.00008%, t R 30.2 min). Separation of fraction I-2-e by HPLC (Nacalai tesque Cholester 10 × 250 mm, 45% MeCN(aq) at 2.0 mL/min, UV detection at 210 nm) yielded 2 (5.8 mg, 0.00041%, t R 51.0 min). Separation of fraction I-2-h by HPLC (Nacalai tesque Cholester 10 × 250 mm, 55% MeCN(aq) at 2.0 mL/min, UV detection at 210 nm) yielded 3 (1.4 mg, 0.0001%, t R 31.2 min) and 4 (6.0 mg, 0.00042%, t R 34.1 min).
Ceramicine M (1)
White amorphous solid. [α] 30D −47° (c 1.0, CHCl3). IR (film) ν max cm−1: 3350 and 1690. UV λ max (MeOH) nm (log ε): 204 (4.04). CD λ max (MeOH) nm (Δε): 295 (+0.027), 225 (−1.2), 222 (+1.8), and 203 (−7.6). 1H and 13C NMR, see Table 1. ESIMS m/z 419 (M + Na)+. HRESIMS m/z 419.1823 [calcd. for C24H28NaO5 (M + Na)+: 419.1834].
Ceramicine N (2)
White amorphous solid. [α] 31D +50° (c 1.0, CHCl3). IR (film) ν max cm−1: 3480 and 1680. UV λ max (MeOH) nm (log ε): 217 (3.74). CD λ max (MeOH) nm (Δε): 336 (−0.46), 217 (+5.0), 208 (+4.5), and 205 (+4.6) nm. 1H and 13C NMR, see Table 2. ESIMS m/z 447 (M + Na)+. HRESIMS m/z 447.2157 [calcd. for C26H32NaO5 (M + Na)+: 447.2147].
Ceramicine O (3)
White amorphous solid. [α] 30D +10° (c 1.0, CHCl3). IR (film) ν max cm−1: 3480, 1730, and 1680. UV λ max (MeOH) nm (log ε): 204 (4.30). CD λ max (MeOH) nm (Δε): 300 (−1.9), 222 (+10.6), and 205 (+4.9). 1H and 13C NMR, see Table 3. ESIMS m/z 429 (M + Na)+. HRESIMS m/z 429.2045 [calcd. for C26H30NaO4 (M + Na)+: 429.2042].
Ceramicine P (4)
White amorphous solid. [α] 31D +34° (c 1.0, CHCl3). IR (film) ν max cm−1: 3480, 1740, and 1690. UV λ max (MeOH) nm (log ε): 204 (3.99). CD λ max (MeOH) nm (Δε): 336 (−1.3) and 212 (+9.5). 1H and 13C NMR, see Table 4. ESIMS m/z 475 (M + Na)+. HRESIMS m/z 475.2111 [calcd. for C27H32NaO6 (M + Na)+: 475.2097].
13C NMR chemical shift calculations
The conformations were obtained using Monte Carlo analysis with MMFF94 force field [10,11,12,13] and charges on Macromodel 9.1 [14]. Geometries were further optimized by using RI-J [15,16,17] approximation at the DFT [18] B3LYP/TZVP [17, 19,20,21] level of theory. NMR shielding constant calculations were performed on the optimized ground state geometries at the DFT B3LYP/TZVP level of theory. All DFT calculations were performed using Turbomole 7.0 [22]. The 13C NMR chemical shifts of the isomers were obtained by Boltzmann averaging the 13C NMR chemical shifts of the stable conformers.
Synthesis of 6
Compound 5 (40 mg, 0.1 mmol) was dissolved in 8 mL of THF, put under argon, and cooled to 0 °C. To the solution of 5, NaH (76 mg, 3.2 mmol) was added and the solution was stirred. After 2 h, CS2 (800 µL, 13.2 mmol) was added, and the solution was further stirred. After 1.5 h, the solution was returned to rt before adding MeI (400 µL, 6.4 mmol) and stirred for another 4 h. Finally, cold water was added to the reaction mixture and partitioned with Et2O. The Et2O layer was dried over anhydrous Na2SO4 before being dried under reduced pressure to afford solid residues. The residues were separated by silica gel column chromatography (n-hexane/EtOAc, 10:1 → 3:1) to afford 15 (30 mg, 62%).
To a solution of 15 (24 mg, 0.048 mmol) in toluene (2.0 mL) under argon at rt, tris(trimethylsilyl)silane (18 µL, 0.058 mmol) and V-40 (1.2 mg, 0.0048 mmol) was added. The mixture was then stirred for 2 h at 90 °C. The reaction mixture was dried under reduced pressure to afford solid residues. The residues were separated by silica gel column chromatography (n-hexane/EtOAc, 10:1 → 3:1) to afford 6 (4.9 mg, 26%).
Compound 6
White amorphous solid. [α] 19D +115° (c 0.5, CHCl3). IR (film) ν max cm−1: 1680. UV λ max (MeOH) nm (log ε): 204 (4.02). CD λ max (MeOH) nm (Δε): 334 (−0.62), 218 (+9.5), and 203 (+5.1). 1H NMR (400 MHz, CDCl3) δ: 0.80 (3H, s), 1.14 (3H, s), 1.18 (3H, s), 1.35 (3H, s), 1.40 (1H, t, 11.8), 1.63 (1H, m), 1.80 (1H, m), 1.88 (1H, m), 1.92 (1H, d, 11.8), 2.06 (1H, dd, 11.5, 6.3), 2.32 (1H, ddd, 15.2, 7.3, 3.5), 2.45 (1H, m), 2.49 (1H, m), 2.53 (1H, dd, 11.8, 4.5), 2.82 (1H, dd, 10.7, 7.4), 3.62 (1H, d, 7.1), 3.74 (1H, d, 7.1), 4.09 (1H, td, 11.8, 4.5), 5.49 (1H, br s), 5.82 (1H, d, 9.7), 6.29 (1H, s), 6.95 (1H, d, 9.7), 7.24 (1H, s), 7.37 (1H, s). 13C NMR (100 MHz, CDCl3) δ: 14.5, 18.5, 20.6, 21.8, 27.5, 33, 34.2, 41.2, 42.2, 42.3, 45.9, 47.2, 47.4, 51.9, 58.5, 71.8, 79.1, 111.1, 119.2, 124.8, 129.9, 139.7, 142.5, 151.3, 162.9, 203.2. HRESIMS m/z 415.2241 [calcd. for C26H32NaO3 (M + Na)+: 415.2249].
Synthesis of 3
Compound 5 (100 mg, 0.25 mmol) was dissolved in 10 mL of CH2Cl2, put under argon, and cooled to 0 °C. To the solution of 5, PCC (181 mg, 0.84 mmol) was added, and the solution was stirred at rt. After 2 h, more PCC (125 mg, 0.58 mmol) was added, and the solution was further stirred at rt for 2 h. To the resulting mixture, diethyl ether was added, and the solids were filtered through Celite. The filtrates were then dried under reduced pressure to afford solid residues. The residues were separated by silica gel column chromatography (n-hexane/EtOAc = 1:1) to afford 3 (70 mg, 70%).
Preparation of iBu2AlOiPr
iBu2AlH (1.0 M in toluene, 5.0 mmol, 5.0 mL) was put under argon and cooled to 0 °C. Isopropanol (390 µL, 5.0 mmol) was then added, and the mixture was stirred for 1 h at rt before being used in the reactions below.
Preparation of iBu2AlOH
iBu2AlH (1.0 M in toluene, 5.0 mmol, 5.0 mL) was put under argon and cooled to 0 °C. Water (90 µL, 5.0 mmol) was then added, and the mixture was stirred for 1 h at rt before being used in the reaction below.
Table 4: entry 1
To a solution of 3 (5 mg, 0.012 mmol) in toluene (0.5 mL) under argon, iBuAlOiPr (72 µL, 0.072 mmol) was added at rt. The mixture was then stirred at 70 °C for 5 h, and cooled to rt before adding 72 µL of water and stirred for another hour. To the resulting mixture, EtOAc was added, and the solids were filtered through Celite. The filtrates were then dried under reduced pressure to afford solid residues. The residues were separated by ODS HPLC (Shiseido ODS MGII 4.6 × 250 mm, MeOH/H2O, 75:25 at 0.5 mL/min, UV detection at 210 nm) to afford 5 (1.7 mg, 33%, t R = 28.0 min) and 7 (0.15 mg, 3%, t R = 26.4 min).
Table 4: entry 2
To a solution of 3 (5 mg, 0.012 mmol) in toluene (0.5 mL) under argon, iBuAlOiPr (24 µL, 0.024 mmol) was added at rt. The mixture was then refluxed for 0.5 h, and cooled to rt before adding 72 µL of water and stirred for another hour. To the resulting mixture, EtOAc was added, and the solids were filtered through Celite. The filtrates were then dried under reduced pressure to afford solid residues. The residues were separated by ODS HPLC (Shiseido ODS MGII 4.6 × 250 mm, MeOH/H2O, 75:25 at 0.5 mL/min, UV detection at 210 nm) to afford 5 (1.8 mg, 36%, t R = 28.0 min), 7 (0.6 mg, 12%, t R = 26.4 min), and 8 (0.4 mg, 8%, t R = 29.6 min).
Table 4: entry 3
To a solution of 3 (20 mg, 0.049 mmol) in toluene (2.0 mL) under argon, iBuAlOH (390 µL, 0.39 mmol) was added and stirred at rt. After 20 h, the mixture was cooled to 0 °C before adding 1 N HCl (5 mL). The resulting mixture was then partitioned with EtOAc, and the EtOAc layer was dried with anhydrous Na2SO4 before being dried under reduced pressure to afford solid residues. The residues were separated by preparative TLC (benzene/EtOAc, 4:1) to afford 5 (7.2 mg, 36%) and 7 (3.0 mg, 15%).
Compound 7
White amorphous solid. [α] 20D +43° (c 0.1, CHCl3). IR (film) ν max cm−1: 3730 and 1680. UV λ max (MeOH) nm (log ε): 203.5 (3.95). CD λ max (MeOH) nm (Δε): 341 (−0.59), 219 (+6.0), and 206 (+3.7). 1H NMR (400 MHz, CDCl3) δ: 0.83 (3H, s), 1.18 (3H, s), 1.19 (3H, s), 1.34 (3H, s), 1.63 (1H, m), 1.76 (1H, m), 1.82 (1H, m), 2.01 (1H, d, 12.0), 2.40 (2H, m), 2.53 (1H, m), 2.88 (1H, m), 3.43 (1H, dd, 8.0, 3.2), 3.63 (1H, d, 8.0), 3.79 (1H, d, 8.0), 4.07 (1H, dt, 12.0, 8.0), 5.85 (1H, d, 11.0), 5.92 (1H, br s), 6.30 (1H, s), 6.94 (1H, d, 11.0), 7.25 (1H, s), 7.37 (1H, s). 13C NMR (100 MHz, CDCl3) δ: 14.8, 19.7, 19.8, 20.0, 24.7, 34.6, 37.1, 42.3, 45.9, 46.3, 47.3, 47.6, 51.7, 55.2, 77.6, 79.4, 80.9, 111.0, 124.5, 125.1, 129.8, 139.7, 142.6, 151.2, 158.1, 202.6. HRESIMS m/z 431.2192 [calcd. for C26H32NaO4 (M + Na)+: 431.2198].
Compound 8
White amorphous solid. [α] 22D +58° (c 0.1, CHCl3); IR (film) ν max cm−1: 3730 and 1680. UV λ max (MeOH) nm (log ε): 204.5 (3.85). CD λ max (MeOH) nm (Δε): 336 (−0.44), 289 (−0.11), 243 (−0.49), and 202 (+5.3). 1H NMR (400 MHz, CDCl3) δ: 0.94 (3H, s), 1.09 (3H, s), 1.23 (3H, s), 1.27 (3H, s), 1.73 (1H, m), 1.94 (1H, m), 2.14 (1H, m), 2.33 (1H, m), 2.83 (1H, m), 2.5 (1H, m), 2.61 (1H, d, 12.0), 2.84 (1H, m), 3.42 (1H, d, 12.0), 3.69 (1H, d, 12.0), 3.75 (1H, dd, 12.0, 3.2), 4.45 (1H, dd, 12.0, 3.2), 5.8 (1H, br s), 5.89 (1H, d, 10.0), 6.28 (1H, s), 6.46 (1H, d, 10.0), 7.25 (1H, s), 7.38 (1H, s). 13C NMR (100 MHz, CDCl3) δ: 16.0, 16.4, 16.6, 23.4, 32.8, 33.0, 34.3, 43.2, 44.5, 44.6, 49.7, 49.9, 50.4, 52.7, 61.4, 70.3, 111.3, 120.3, 127.8, 132.5, 140.2, 142.9, 156.2, 162.5, 207.6. HRESIMS m/z 433.2567 [calcd. For C26H32NaO4 (M + Na)+: 433.2555].
Synthesis of 9
To a solution of 6 (3.0 mg, 0.007 mmol) in pyridine (0.5 mL) under argon at rt, acetic acid anhydride (1.3 µL, 0.014 mmol) and 4-dimethylaminopyridine (0.085 mg, 0.0007 mmol) were added. The mixture was then stirred for 15 min and dried under reduced pressure to afford solid residues. The residues were separated by preparative TLC (benzene/EtOAc, 4:1) to afford 9 (1.8 mg, 64%).
Compound 9
White amorphous solid. [α] 21D +28° (c 0.1, CHCl3). IR (film) ν max cm−1: 1740 and 1680. UV λ max (MeOH) nm (log ε): 203.5 (4.78). λ max (MeOH) nm (Δε): 334 (−4.3) and 223 (+43). 1H NMR (400 MHz, CDCl3) δ: 0.86 (3H, s), 1.19 (3H, s), 1.25 (3H, s), 1.33 (3H, s), 1.62 (1H, m), 1.76 (1H, m), 1.84 (1H, m), 2.11 (1H, d, 12.0), 2.15 (3H, s), 2.32 (2H, m), 2.54 (1H, m), 2.84 (1H, m), 3.67 (1H, d, 7.2), 3.75 (1H, d, 7.2), 4.20 (1H, dd, 12.0, 8.8), 4.92 (1H, d, 8.8), 5.55 (1H, br s), 5.85 (1H, d, 9.6), 6.29 (1H, s), 6.93 (1H, d, 9.6), 7.22 (1H, s), 7.36 (1H, s). 13C NMR (100 MHz, CDCl3) δ: 15.1, 19.9, 20.0, 20.4, 21.5, 24.9, 34.8, 37.9, 42.2, 45.9, 46.7, 47.6, 51.8, 55.5, 75.3, 79.6, 80.7, 111.2, 124.4, 125.5, 129.9, 139.8, 142.7, 151.3, 156.0, 170.3, 202.3. HRESIMS m/z 473.2313 [calcd. for C28H34NaO5 (M + Na)+: 473.2304].
Synthesis of 10
To a solution of 6 (3.0 mg, 0.007 mmol) in pyridine (0.5 mL) under argon at rt, benzoyl chloride (1.7 µL, 0.015 mmol) and 4-dimethylaminopyridine (0.085 mg, 0.0007 mmol) were added. The mixture was then stirred for 2 h and dried under reduced pressure to afford solid residues. The residues were separated by preparative TLC (benzene/EtOAc, 4:1) to afford 10 (1.1 mg, 32%).
Compound 10
White amorphous solid. [α] 19D +81° (c 0.1, CHCl3). IR (Film) ν max cm−1: 1720 and 1680. UV λ max (MeOH) nm (log ε): 225 (4.19) and 201.5 (4.20). CD λ max (MeOH) nm (Δε): 329 (−0.81), 227 (+11), and 211 (+6.2). 1H NMR (400 MHz, CDCl3) δ: 0.91 (3H, s), 1.24 (3H, s), 1.35 (3H, s), 1.40 (3H, s), 1.65 (1H, m), 1.75 (1H, m), 1.84 (1H, m), 2.20 (1H, d, 12.0), 2.22 (2H, m), 2.58 (1H, m), 2.82 (1H, m), 3.71 (1H, d, 7.6), 3.76 (1H, d, 7.6), 4.34 (1H, dd, 12.0, 9.6), 5.21 (1H, d, 9.6), 5.49 (1H, br s), 5.87 (1H, d, 10.0), 6.28 (1H, s), 6.96 (1H, d, 10.0), 7.20 (1H, s), 7.35 (1H, s), 7.47 (2H, t, 7.6), 7.58 (1H, t, 7.6), 8.14 (2H, d, 7.6); 13C NMR (100 MHz, CDCl3) δ: 14.9, 20.0, 20.9, 24.7, 37.4, 42.3, 46.0, 47.6, 51.6, 55.6, 75.3, 79.6, 81.2, 111.0, 124.5, 125.7, 128.6, 130.0, 133.2, 139.8, 142.7, 151.2, 156.1, 165.7, 185.2, 202.4. HRESIMS m/z 535.2452 [calcd. for C33H36NaO5 (M + Na)+: 535.2460].
Synthesis of 11
To a solution of 6 (1.5 mg, 0.004 mmol) in DMF (0.5 mL) under argon at rt, EtI (5.7 µL, 0.018 mmol) and NaH (0.4 mg, 0.018 mmol) were added, and the mixture was then stirred. After 4 h, saturated NH4Cl was added, and the resulting mixture was then partitioned with EtOAc. The EtOAc layer was dried with anhydrous Na2SO4 before being dried under reduced pressure to afford solid residues. The residues were separated by preparative TLC (benzene/EtOAc, 4:1) to afford 11 (1.3 mg, 75%).
Compound 11
White amorphous solid. [α] 22D +31° (c 0.1, CHCl3). IR (Film) ν max cm−1: 1680. UV λ max (MeOH) nm (log ε): 203.5 (3.70). CD λ max (MeOH) nm (Δε): 332 (−0.25), 224 (+2.4), and 202 (+1.2). 1H NMR (400 MHz, CDCl3) δ: 0.84 (3H, s), 1.16 (3H, s), 1.19 (3H, s), 1.24 (3H, t, 6.8), 1.32 (3H, s), 1.66 (1H, m), 1.78 (1H, m), 2.38 (2H, m), 2.46 (1H, m), 2.76 (1H, d, 12.0), 2.84 (1H, m), 3.54 (2H, q, 6.8), 3.60 (1H, d, 7.2), 3.76 (1H, d, 7.2), 4.10 (1H, dd, 12.0, 2.8), 5.83 (1H, d, 10.0), 5.98 (1H, br s), 6.31 (1H, s), 6.94 (1H, d, 10.0), 7.25 (1H, s), 7.37 (1H, s). 13C NMR (100 MHz, CDCl3) δ: 14.7, 15.6, 19.6, 20.2, 20.8, 24.3, 34.7, 36.4, 41.9, 45.5, 46.2, 46.5, 47.5, 51.7, 55.3, 67.6, 78.0, 79.1, 88.7, 111.1, 124.9, 129.8, 139.6, 142.5, 151.3, 157.4, 202.8. HRESIMS m/z 459.2514 [calcd. for C28H36NaO4 (M + Na)+: 459.2511].
LDA inhibitory activity and cytotoxicity
MC3T3-G2/PA6 murine pre-adipocytes (Riken Cell Bank, Ibaraki, Japan) were maintained in basal medium [alpha minimum essential medium (α-MEM) (Wako, Osaka, Japan) supplemented with 10% FBS (Cell Culture Bioscience, Tokyo, Japan)]. LDA inhibitory activity and cytotoxicity on MC3T3-G2/PA6 were measured using the same methods as in our previous report [5, 6]. Briefly, the LDA inhibitory activity of the samples was measured on the basis of the amount of LDA after 6 days of incubation with a mixture of 3-isobutyl-1-methylxanthine (IBMX), dexamethasone (DEX), and insulin (MDI inducer), and was expressed as IC50 value (the concentration of the sample causing 50% inhibition of LDA relative to an untreated control). The cytotoxicity was evaluated indirectly via MTT assay which is based on mitochondrial succinate dehydrogenase activity and confirmed via microscopic observation. The cytotoxicity was expressed as CC50 value which was defined as the concentration of the sample causing 50% cell viabilities relative to an untreated control.
Change history
03 April 2019
The article Ceramicines M���P from Chisocheton ceramicus: isolation and structure���activity relationship study.
References
Mohamad K, Hirasawa Y, Lim CS, Awang K, Hadi AHA, Takeya K, Morita H (2008) Ceramicine A and walsogyne A, novel limonoids from two species of Meliaceae. Tetrahedron Lett 49:4276–4278
Mohamad K, Hirasawa Y, Litaudon M, Awang K, Hadi AHA, Takeya K, Ekasari W, Widyawaruyanti A, Zaini NC, Morita H (2009) Ceramicines B–D, new antiplasmodial limonoids from Chisocheton ceramicus. Bioorg Med Chem 17:727–730
Wong CP, Shimada M, Nagakura Y, Nugroho AE, Hirasawa Y, Kaneda T, Awang K, Hadi AHA, Mohamad K, Shiro M, Morita H (2011) Ceramicines E–I, new limonoids from Chisocheton ceramicus. Chem Pharm Bull 59:407–411
Wong C, Shimada M, Nugroho AE, Hirasawa Y, Kaneda T, Hadi AHA, Osamu S, Morita H (2012) Ceramicines J–L, new limonoids from Chisocheton ceramicus. J Nat Med 66:566–570
Wong CP, Deguchi J, Nugroho AE, Kaneda T, Hadi AHA, Morita H (2013) Ceramicines from Chisocheton ceramicus as lipid-droplets accumulation inhibitors. Bioorg Med Chem Lett 23:1786–1788
Wong CP, Kaneda T, Hadi AHA, Morita H (2014) Ceramicine B, a limonoid with anti-lipid droplets accumulation activity from Chisocheton ceramicus. J Nat Med 68:22–30
Iijima C, Wong CP, Nugroho AE, Sotozono Y, Someya S, Hirasawa Y, Kaneda T, Hadi AHA, Morita H (2016) Anti-melanin deposition activity of ceramicines from Chisocheton ceramicus. J Nat Med 70:702–707
Perchyonok VT (2006) On the use of (TMS)3CH as novel tin-free radical reducing agent. Tetrahedron Lett 47:5163–5165
Bahia PS, Jones MA, Snaith JS (2004) Al-isopropoxydiisobutylalane: a study of the effect of solvent on the rate and stereoselectivity of cyclic ketone reduction. J Org Chem 69:9289–9291
Halgren TA (1990) Maximally diagonal force constants in dependent angle-bending coordinates. II. Implications for the design of empirical force fields. J Am Chem Soc 112:4710–4723
Halgren TA (1992) The representation of van der Waals (vdW) interactions in molecular mechanics force fields: potential form, combination rules, and vdW parameters. J Am Chem Soc 114:7827–7843
Halgren TA (1996) Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem 17:490–519
Halgren TA (1996) Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J Comput Chem 17:520–552
Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC (1990) Macromodel—an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem 11:440–467
Eichkorn K, Treutler O, Ohm H, Haser M, Ahlrichs R (1995) Auxiliary basis sets to approximate Coulomb potentials. Chem Phys Lett 240:283–289
Eichkorn K, Weigend F, Treutler O, Ahlrichs R (1997) Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor Chem Acc 97:119–124
Weigend F (2006) Accurate Coulomb-fitting basis sets for H to Rn. Phys Chem Chem Phys 8:1057–1065
Treutler O, Ahlrichs R (1995) Efficient molecular numerical integration schemes. J Chem Phys 102:346–354
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098–3100
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Weigend F, Häser M, Patzelt H, Ahlrichs R (1998) RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem Phys Lett 294:143–152
TURBOMOLE V7.0 (2015) A development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007; TURBOMOLE GmbH, since 2007. http://www.turbomole.com. Accessed 13 July 2017
Acknowledgements
This work was partly supported by Japan Society for the Promotion of Science KAKENHI (JP 16K08309), Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original version of this article was revised due to a retrospective Open Access order
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Nugroho, A.E., Hashimoto, A., Wong, CP. et al. Ceramicines M–P from Chisocheton ceramicus: isolation and structure–activity relationship study. J Nat Med 72, 64–72 (2018). https://doi.org/10.1007/s11418-017-1109-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-017-1109-2