
Abstract
Epithelial to mesenchymal transition (EMT) occurs during embryogenesis or under pathological conditions such as hypoxia, injury, chronic inflammation, or tissue fibrosis. In renal tubular epithelial cells (MDCK), TGF-β1 induces EMT by reducing or increasing epithelial or mesenchymal marker expression, respectively. In this study, we confirmed that the cAMP analogues, 8-CPT-cAMP or N6-Ph-cAMP, inhibited the TGF-β1-driven overexpression of the mesenchymal markers ZEB-1, Slug, Fibronectin, and α-SMA. Furthermore, we showed that A1, A2A, P2Y1, P2Y11, and P2X7 purine receptor agonists modulated the TGF-β1-induced EMT through the involvement of PKA and/or MAPK/ERK signaling. The stimulation of A2A receptor reduced the overexpression of the EMT-related markers, mainly through the cAMP-dependent PKA pathway, as confirmed by cell pre-treatment with Myr-PKI. Both A1 and P2Y1 receptor stimulation exacerbated the TGF-β1-driven effects, which were reduced by cell pre-treatment with the MAPK inhibitor PD98059, according to the increased ERK1/2 phosphorylation upon receptor activation. The effects induced by P2Y11 receptor activation were oppositely modulated by PKA or MAPK inhibition, in line with the dual nature of the Gs- and Gq-coupled receptor. Differently, P2X7 receptor induced, per se, similar and not additive effects compared to TGF-β1, after prolonged cell exposure to BzATP. These results suggest a putative role of purine receptors as target for anti-fibrotic agents.
Similar content being viewed by others


Introduction
The epithelial to mesenchymal transition (EMT) is a fundamental process of the embryogenesis and morphogenesis, wherein epithelial cells gradually lose their characteristic features and functionality thus acquiring a mesenchymal cell phenotype [1]. EMT reactivation in adults seems to play a pivotal role in tissue repair (wound healing and/or scar formation) through the production of proinflammatory cytokines. Therefore, it has been considered as a crucial link between inflammation and the progression of organ fibrosis and cancer [2].
Although the contribution of EMT to tissue fibrosis in vivo is not univocal and it should be better defined [3,4,5,6], the proinflammatory cytokine TGF-β1 has been widely considered as one of the most potent activators of the experimentally induced EMT for its profibrotic effect in many cell types. Upon the binding to its receptor, TGF-β1 is able to induce EMT either via canonical pathways involving Smad2/3/4 phoshorylation and the transcription of ZEB1–2, Snail or Slug [7, 8], or via non-Smad mechanisms such as PI3K/Akt/mTOR [9, 10], RHO-GTP-ase, ERK, p38, and JUN N-terminal kinase (JNK) MAPK pathways [11, 12].
The most notable hallmark of EMT is the decrease of E-cadherin expression and, as consequence, the loss of epithelial cell-cell contacts and, in parallel, the increased expression of mesenchymal markers such as N-cadherin, Vimentin, and α-SMA (the actin isoform specific to myofibroblasts) as well as the production of matrix proteins (e.g., Fibronectin and collagen) [13, 14]. EMT features and new insights are extensively reviewed in “EMT: 2016” by A. Nieto et al. [15].
It is well known that cyclic AMP (cAMP) system regulates the function/activity of many hormones, some of which modulate the extracellular matrix composition. Cyclic AMP modulates key functions of fibroblasts, including those induced by TGF-β1. Indeed, the increase of cAMP intracellular levels, induced by forskolin or phosphodyesterase inhibitors, as well as the administration of dibutyryl cAMP prevented TGF-β1-induced Smad-specific gene transactivation and antagonized the effects of TGF-β1 on the expression of extracellular matrix components (collagen, growth factors of the connective tissue, and metalloproteinase-1) [16]. In addition to protein kinase A (PKA), exchange proteins directly activated by cAMP (Epac) were identified as regulators of TGF-β1-driven EMT [17, 18]. These proteins, acting in a PKA-independent manner, represent a collateral pathway in the cAMP signaling. However, PKA and Epac result to be often associated with the same biological process, wherein they fulfill either synergistic or opposite effects [19].
Besides the well-described role of cAMP, we took advantage of the involvement of MAPK pathway in TGF-β1-induced EMT to investigate its role in the regulation of fibrotic processes.
In this context, it has been observed that, similarly to TGF-β1, H2O2 promotes either the phosphorylation of Smad2, p38 MAPK, and ERK1/2, or the overexpression of α-SMA and Fibronectin, whereas it decreases the expression of E-cadherin in renal tubular epithelial cells [20]. Furthermore, other groups reported that (i) phosphoglucose isomerase/autocrine motility factor (PGI/AMF) activates the EMT in breast cancer cells, thus favoring their metastatic invasion [21]; (ii) aldosterone induces EMT via mitochondrial-derived ROS [22]; (iii) angiotensin II plays a key role in the high-glucose-induced upregulation of Fibronectin expression in human peritoneal mesothelial cells implicated in peritoneal fibrosis [23]; (iv) nitric oxide attenuates the TGF-β1-induced transition of alveolar epithelial cells to myofibroblasts, both in vitro and in vivo [24]; and (v) prostacyclin analogues inhibit collagen expression in cultured cardiac fibroblasts by increasing cAMP formation [25]. Cyclic AMP and MAPK signaling pathways also derives from the stimulation of a plethora of metabotropic receptors that might play a crucial role in EMT/fibrosis. Among these receptors, there are few studies regarding the role of purine receptors in modulating this process. It has been reported that MDCK cells express a large number of purine receptor subtypes, mainly belonging to the P2 family, both P2X and P2Y. Among them, P2Y subtypes (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, and P2Y14) result to be the most expressed [26,27,28], whereas there is less evidence of P1 adenosine receptor expression on these cells [29,30,31].
Therefore, in the present study, we investigated the role of some prototypal P1 (A1, A2A) and P2 (P2Y1, P2Y11, P2X7) purine receptors in TGF-β1-driven EMT in MDCK cells by focusing on the underlying mechanisms linked to their anti-fibrotic effects.
Materials and methods
Reagents
Recombinant human transforming growth factor-β1 (TGF-β1) was purchased from Millipore (Milan, Italy). PD98059, 3-isobutyl-1-methylxanthine (IBMX), MRS2365, MRS2179, 2-chloro-N6-cyclopentyladenosine (CCPA), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), 2′,3′-(benzoyl-4-benzoyl)-ATP (BzATP), and periodate-oxidized ATP (OxATP) were purchased from Sigma-Aldrich. NF546 (4,4′-(carbonylbis(imino-3,1-phenylene-carbonylimino-3,1-(4-methyl-phenylene)carbonylimino))-bis(1,3-xylene-α,α′-diphosphonic acid tetrasodium salt), NF340 [4,4′-(carbonylbis(imino-3,1-(4-methyl-phenylene)carbonylimino))bis(naphthalene-2,6-disulfonic acid) tetrasodium salt], CGS21680, ZM241385 (4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol), and 8-CPT-2Me-cAMP (8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate sodium salt) were purchased from Tocris Bioscience (Ellisville, MO, USA). Myr-PKI (protein kinase inhibitor (14-22)-amide myristoylated) was purchased from Enzo Life Sciences (Roma, Italy). 8-CPT-cAMP (8-(4-chlorophenylthio)adenosine-3′,5′-cyclic monophosphate) and N6-Ph-cAMP (N6-phenyladenosine-3′,5′-cyclic monophosphate) were purchased from Biolog Life Science Institute (Bremen, Germany). Alexa Fluor 594 phalloidin, 4′,6-diamino-2-phenyl-indole (DAPI), and Slowfade Antifade Reagent were purchased from Life Technologies.
Cell culture
Madin Darby canine kidney (MDCK) cells were purchased from Sigma (Sigma-Aldrich, Milan, Italy) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Life Technologies, Monza, Italy) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco), 100 I.U. Penicillin, and 100 μg/mL Streptomycin (Gibco), then incubated in a humidified chamber with 5% CO2, at 37 °C. For all treatments, cells were grown to sub-confluency and subjected to immunofluorescence study, Western blot and RT-PCR analyses as follows.
Western blot
In order to analyze the expression of the major epithelial and mesenchymal markers, Western blot analyses were performed on MDCK cells. MDCK cells were seeded overnight onto 100 × 20-mm culture dishes (BD Falcon) at 500,000 cells/dish in 6 mL of DMEM supplemented with 10% heat-inactivated FBS, 100 I.U. Penicillin, and 100 μg/mL Streptomycin per dish. After 24-h starvation, cells were submitted to different treatments in DMEM supplemented with 0.5% FBS and 1% Penicillin/Streptomycin. After treatment, cells were washed twice with 1× PBS (Sigma-Aldrich), lysed with RIPA Buffer (Sigma-Aldrich) supplemented with 1% Protease Inhibitor Cocktail (Sigma-Aldrich), scraped off, and clarified by centrifugation at 12,500×g for 20 min. Before performing Western blot, a sample buffer (5× Laemmli buffer with 10% mercaptoethanol) was added to melted lysates 1:4. Protein concentrations were obtained using the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA) based on the Bradford method. An equal amount of 50–70 μg of protein was resolved by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The resolved proteins were transferred onto a nitrocellulose membrane and then incubated with blocking buffer 1× PBS containing 0.1% Tween-20 (PBST) and 5% non-fat dry milk for 2 h, RT, and subsequently incubated with specific primary antibody at 4 °C, overnight. After washing with PBST, the membrane was further incubated with corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies at RT for 1 h. Membranes were finally washed, before subjecting them to ECL Plus Western Blotting Detection Reagent (Amersham, GE Healthcare). The immunoreactive bands were visualized under a chemiluminescence detection system (UVItec, Cambridge, UK). Band intensity data were obtained using Quantity One software (Bio-Rad Laboratories). Blotting membranes were stripped and re-probed with anti-actin antibody as equal loading control. The following primary antibodies were used: anti-E-cadherin (BD Biosciences, San Jose, CA, USA), anti-N-cadherin and β-actin (Cell Signaling, Danvers, MA, USA), anti-actin α-smooth muscle (α-SMA), and anti-human Fibronectin (Sigma-Aldrich, Saint Louis, MO, USA). Secondary anti-rabbit and anti-mouse IgG and HRP-linked antibodies were purchased from Cell Signaling (Cell Signaling, Danvers, MA, USA). E-cadherin was diluted 1:800 in 2.5% non-fat dry milk/1× PBS/0.1% Tween 20, N-cadherin and β-actin were diluted 1:1000 in 2.5% non-fat dry milk/1× PBS/0.1% Tween 20, α-SMA was diluted 1:1500 in 1% non-fat dry milk/1× PBS/0.1% Tween 20, and Fibronectin was diluted 1:2000 in 5% non-fat dry milk/1× PBS/0.1% Tween 20. A secondary anti-rabbit IgG, HRP-linked antibody was used for N-cadherin and Fibronectin, whilst a secondary anti-mouse IgG, HRP-linked antibody was used for E-cadherin, α-SMA, and β-actin. All secondary antibodies were used at 1:2500 dilution in 2.5% non-fat dry milk/1× PBS/0.1% Tween 20 or 2.5% BSA/1× PBS/0.1% Tween 20.
Real-time PCR
To evaluate mesenchymal transcriptional characteristics of MDCK cells, real-time PCR (RT-PCR) of EMT-related markers was performed. MDCK cells were grown in 24-well plates into medium supplemented with 0.1% FBS and 100 I.U. Penicillin and 100 μg/mL Streptomycin. After 24-h starvation, cells were incubated with (1) TGF-β1 alone up to 72 h; (2) N6-Ph-cAMP, 8-CPT-cAMP, 8-CPT-2Me-cAMP, and IBMX alone or in association with TGF-β1 for 48 h; (3) CGS21680, CCPA, MRS2365, and NF546 in association with TGF-β1 for 48 h; (4) Myr-PKI or PD98059 30 min before TGF-β1/cAMP analogue co-treatment until the end of experiment; (5) ZM241385, DPCPX, MRS2179, and NF340 30 min before TGF-β1/P1 or P2 agonists co-treatment until the end of experiment; (6) BzATP up to 96 h; and (7) OxATP or PD98059 30 min before BzATP treatment until the end of experiment. RNA was extracted from MDCK cells by using Qiagen RNeasy Plus Mini Kit according to manufacturer’s instructions. The forward and reverse primers were purchased from Integrated DNA Technologies (IDT) (Leuven, Belgium): ZEB1 forward 5′-CAA GGT GGC CAT TCT GTT AT-3′ and reverse 5′-CTA GGC TGC TCA AGA CTG TAG-3′, E-cadherin forward 5′-AAG CGG CCT CTA CAA CTT CA-3′ and reverse 5′-AAC TGG GAA ATG TGA GCA CC-3′, N-cadherin forward 5′-CAA CTT GCC AGA AAA CTC CAG G-3′ and reverse 5′-ATG AAA CCG GGC TAT CAG CTC-3′, Vimentin forward 5′-GCC ATC AAC ACC GAG TTC AA-3′ and reverse 5′-GGA AGC GCA CCT TGT CGA T-3′, Slug forward 5′-CGT TTT CCA GAC CCT GGT TA-3′ and reverse 5′-TGA CCT GTC TGC AAA TGC TC-3′, Fibronectin forward 5′-GCA ACT CTG TGG ACC AAG G-3′ and reverse 5′-CAC TGG CAC GAG AGC TTA AA-3′, α-SMA forward 5′-TGT TCC AGC CGT CCT TCA T-3′ and reverse 5′-GGC GTA GTC TTT CCT GAT G-3′, and GAPDH forward 5′-CAT CAC TGC CAC CCA GAA G-3′ and reverse 5′-CAG TGA GCT TCC CGT TCA G-3′. The PCR reaction comprised pre-denaturation at 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control to normalize RT-PCR readout and analyzed using the 2−ΔΔCt method of Livak and Schmittgen.
Immunofluorescence stainings
To probe changes in MDCK cell morphology after TGF-β1 stimulation, the cells were grown on glass coverslips in 24-multiwell plates (Falcon) at 20 × 103 cells/well. The cells were challenged at 24, 48, and 72 h with growing concentrations of TGF-β1 (2.5–5–10 ng/mL). At the end of the exposure, the cells were processed for fluorescence staining of F-actin. Briefly, cells were washed twice with 1× PBS and then fixed with 4% paraformaldehyde (Sigma) in PBS, at RT, for 30 min and then permeabilized with 0.1% IGEPAL (Sigma) in 1× PBS, at RT, for 10 min. After washing, the cells were incubated with Phalloidin conjugated with Alexa Fluor 594 at a dilution of 1:200 in 1% BSA/1× PBS at 37 °C for 30 min. At the end of incubation, they were washed twice with 1× PBS and were incubated in the fluorescent dye DAPI at 1 μg/mL in PBS to stain the nuclei of cells. After the final washing step, the glass coverslips were mounted onto a slide using Slow Fade and fluorescence images were acquired using a Zeiss confocal microscopy system (LSM510 META; Carl Zeiss, Jena, Germany) equipped with an inverted microscope (Zeiss Axiovert200) and an oil-immersion objective (PlanNeofluar; ×400).
Statistical analysis
Data are presented as means ± standard error of mean (SEM). Statistical significance (P < 0.05) was evaluated by Student’s t test using GraphPad Prism software. All experiments were performed at least three times.
Results
TGF-β1 modifies the MDCK cell morphology and the expression of EMT-related markers
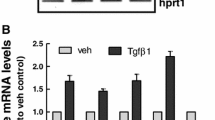
MDCK cells have been often used as a model to evaluate different aspects of EMT [32]. A defining feature of EMT is a change in cell morphology and loss of cell-cell contact. We first determined the optimum concentration and the time of exposure of TGF-β1 able to induce, in MDCK cells, the expression of EMT-related genes, ZEB1, Slug, Fibronectin, and α-SMA, which are associated to profibrotic processes, thus contributing to the loss of epithelial cell polarity and the acquisition of a fibroblastoid phenotype. We evaluated the dose-effect curve (up to 800 pM, corresponding to 20 ng/mL) for 48 h and the time course (up to 72 h) of TGF-β1 by RT-PCR analysis. The maximal effect was obtained using a concentration of 300 pM (corresponding to about 7.5 ng/mL) and the EC50 was about 230 ± 1.9 pM (corresponding to about 5.0 ng/mL) (Fig. 1a, b), already observed after 48 h of cell stimulation (Fig. 1c, d). Phalloidin staining of MDCK cells, exposed to increasing concentrations (2.5, 5, and 10 ng/mL) of TGF-β1 for 48 h, confirmed that cells underwent a marked and dose-dependent morphological change, from cobble to spindle-like morphology with elongated F-actin stress fibers and weaker cell-cell adhesion interactions, a classical feature of mesenchymal cells (Fig. 1e). Therefore, we chose to expose MDCK cells to TGF-β1 5.0 ng/mL for 48 h to pharmacologically modulate the effects on EMT marker expression.

Effect of TGF-β1 on EMT marker expression. a Dose-effect curves of TGF-β1. MDCK cells were challenged with increasing concentrations of TGF-β1 ranging from 0.75 ng/mL (30 pM) to 20 ng/mL (800 pM) for 48 h to examine TGF-β1-mediated changes in ZEB1, Slug expression (left) and α-SMA, Fibronectin (right) by RT-PCR analysis. b Time course of TGF-β1. The effect of TGF-β1 on ZEB1, Slug (left) and α-SMA, Fibronectin (right) expression was evaluated by RT-PCR analysis in MDCK cells exposed to 5 ng/mL (200 pM) TGF-β1 up to 72 h. Each value represents the mean ± SEM of at least three independent experiments, and it is expressed as relative amount of mRNA normalized to GAPDH. c Representative photomicrographs of MDCK cells. Cells were challenged with increasing concentration of TGF-β1 (2.5–10 ng/mL) for 48 h. The sub-confluent monolayers were stained with rhodamine-phalloidin (red) to label F-actin of the cytoskeleton. Scale bars: 10 μm. TGF-β1-stimulated cells showed spindle-like morphology with elongated F-actin stress fibers, indicative of mesenchymal cells
Influence of the downstream effectors of cAMP system on the TGF-β1-induced EMT in MDCK cells
Since cAMP is one of the main intracellular mediators that are able to interfere with TGF-β1 signaling, we evaluated the role of cAMP effectors on TGF-β1-driven EMT [17]. We examined the classic cAMP-dependent effector PKA, as well as Epac proteins that are able to activate Ras superfamily small GTPases Rap1 and Rap2 [33, 34]. We first evaluated whether some stable cAMP analogues or the inhibitor of phosphodiesterase, IBMX, could influence per se the expression of the main EMT-related genes (the transcription factors, ZEB1–2 and Slug, as well as the proteins they transcriptionally regulate, N-cadherin, E-cadherin, Fibronectin, Vimentin, and α-SMA), evaluated by RT-PCR. MDCK cells were exposed for 48 h to the cAMP stable analogue 8-CPT-cAMP (5 μM), the PKA- or Epac-specific cAMP analogues, N6-Ph-cAMP (5 μM) or 8-CPT-2Me-cAMP (5 μM), and to a non-specific inhibitor of phosphodiesterases, IBMX (100 μM), able to increase endogenous cAMP intracellular levels. Surprisingly, we observed that lower concentrations (≤ 0.5 μM) of cAMP analogues upregulated the expression of all markers, whereas higher concentrations (≥ 10 μM) downregulated them (data not shown). Concentration of 5 μM did not affect the expression of the mentioned markers; therefore, we evaluated the influence of the cAMP system on the EMT induced by 5 ng/mL TGF-β1 for 48 h. As expected, RT-PCR analysis showed that TGF-β1 upregulated, at different extent, the expression of all the analyzed markers except that of E-cadherin, which was downregulated. 8-CPT-cAMP or N6-Ph-cAMP significantly reduced all the effects induced by TGF-β1. Similarly, 8-CPT-2Me-cAMP counteracted the effects of TGF-β1 on the expression of α-SMA, Vimentin, N-cadherin, and, slightly, that of E-cadherin, but it did not affect the upregulation of ZEB1, Slug, and Fibronectin. These controversial effects were also caused by 100 μM IBMX (Table 1). These results were confirmed by Western blot analysis of Fibronectin, N-cadherin, E-cadherin, and α-SMA protein levels (Fig. 2a, b).

Effects of cAMP analogues and IBMX on TGF-β1-induced EMT. MDCK cells were exposed to 5 ng/mL TGF-β1 for 48 h in the absence or presence of 5 μM 8-CPT-cAMP, 5 μM N6-Ph-cAMP, 5 μM 8-CPT-2Me-cAMP, or 100 μM IBMX. a Representative Western blot analysis of Fibronectin, E-cadherin (left), α-SMA, and N-cadherin (right) with the respective β-actin as loading control. b Quantitative data of densitometric analysis. Each column represents the mean ± SEM of at least three independent experiments, and it is expressed as relative protein expression normalized to β-actin. Student’s t test: *P < 0.05, **P < 0.02, ***P < 0.01, § P < 0.001 vs. untreated cells (control); # P < 0.05, ## P < 0.02, ### P < 0.01, ⌘ P < 0.001 vs. TGF-β1-treated cells
Based on these findings, we focused our attention on those markers mainly involved in the TGF-β1-induced fibrotic processes, ZEB1, Slug, Fibronectin, and α-SMA (evaluated by RT-PCR analysis), and we used 8-CPT-cAMP or N6-Ph-cAMP to evaluate possible cross talks between cAMP system and other intracellular signaling pathways, such as ERK1/2, which is also known to play a pivotal role among the non-canonical TGF-β1 pathways.
As expected, cell exposure to 5 μM 8-CPT-cAMP (Fig. 3a) or 5 μM N6-Ph-cAMP (Fig. 3b) significantly reduced TGF-β1-induced expression of all markers. Cell treatment with the PKA inhibitor, 5 μM Myr-PKI for 48 h, added to culture medium 30 min before TGF-β1 treatment or co-treatment with TGF-β1/cAMP analogues until the end of the experiment reverted the effect of both cAMP analogues on all analyzed EMT markers induced by TGF-β1. Cell exposure to Myr-PKI in the absence of cAMP analogues did not affect EMT-related gene expression. In cells stimulated by TGF-β1, cell pre-treatment for 30 min with the ERK1/2 inhibitor, 10 μM PD98059, did not reduce 8-CPT-cAMP or N6-Ph-cAMP effects, except for α-SMA expression. Different from Myr-PKI, PD98059 alone was able to reduce TGF-β1 effect in most of the markers, confirming the role of ERK1/2 activation in the EMT process.

Effects of 8-CPT-cAMP and N6-Ph-cAMP on TGF-β1-induced EMT. MDCK cells were challenged to 5 ng/mL TGF-β1 for 48 h in the absence or presence of 5 μM 8-CPT-cAMP (a) and 5 μM N6-Ph-cAMP (b). The expression of EMT-associated genes ZEB1, Slug, Fibronectin, and α-SMA was evaluated by RT-PCR analysis. PKA or ERK 1/2 inhibitors, 5 μM Myr-PKI or 10 μM PD98059, were added to culture medium 30 min before TGF-β1 treatment or co-treatment with TGF-β1/cAMP analogues until the end of the experiment. The relative amount of mRNA is presented as ratio of mRNA to GAPDH. Each column represents the mean ± SEM of at least five independent experiments, and it is expressed as relative amount of mRNA normalized to GAPDH. Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.001 vs. TGF-β1-treated cells; # P < 0.05, ## P < 0.01 vs. TGF-β1/cAMP analogue-treated cells
Modulation of purine receptors on EMT induced by TGF-β1 in MDCK cells
In accordance with the effects observed in cAMP analogues and considering that A2A adenosine receptors (A2AR) activate the cAMP system, the stimulation of these receptor subtypes with the specific agonist CGS21680 (50 nM) strongly decreased the expression of ZEB1, Slug, Fibronectin, and α-SMA enhanced by TGF-β1 (evaluated by RT-PCR analysis). This effect was reverted by cell treatment for 48 h with the selective receptor antagonist, ZM241385 (50 nM), added 30 min before the A2AR stimulation (Fig. 4a). This result may be linked to the increase of intracellular cAMP levels via PKA in response to activation of Gαs protein-coupled A2AR. Indeed, PKA inhibition by 5 μM Myr-PKI significantly reduced CGS21680 protective effect on EMT.

Effects of P1 adenosine receptor agonists on TGF-β1-induced EMT. a MDCK cells were challenged to 5 ng/mL TGF-β1 for 48 h in the absence or presence of the selective A2AR agonist, 50 nM CGS21680, or b the selective A1R agonist, 50 nM CCPA. The expression of ZEB1, Slug (left) and Fibronectin, α-SMA (right) was evaluated by RT-PCR analysis. A2AR or A1R antagonists, 50 nM ZM241385 or 100 nM DPCPX, as well as PKA or ERK 1/2 inhibitors, 5 μM Myr-PKI or 10 μM PD98059, were added to culture medium 30 min before TGF-β1 treatment or co-treatment with TGF-β1/A2A or A1R agonists until the end of the experiment. Each column represents the mean ± SEM of at least five independent experiments, and it is expressed as relative amount of mRNA normalized to GAPDH. Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.001 vs. TGF-β1-treated cells; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. TGF-β1/A2AR or A1R agonist-treated cells. c Representative photomicrographs of MDCK cells. Cells were challenged with 5 ng/mL TGF-β1 in the absence or presence of 50 nM CGS21680 or 50 nM CCPA for 48 h. The sub-confluent monolayers were stained with rhodamine-phalloidin (red) and nuclei were counterstained with DAPI (blue). Scale bars: 10 μm
Since the activation of MAPK-ERK1/2 cascade is considered a non-canonical pathway by which TGF-β1 promotes EMT, we evaluated the involvement of this pathway on A2AR-mediated effects, by pre-treating MDCK cells with the specific ERK1/2 inhibitor, PD98059. Interestingly, 10 μM PD9805 significantly decreased the effect of A2AR stimulation on Slug and Fibronectin expressions induced by TGF-β1 (Fig. 4a). Conversely, A1 adenosine receptor (A1R) stimulation by 50 nM CCPA further increased TGF-β1-induced EMT marker expression in MDCK cells. This finding was confirmed by cell pre-treatment with the selective antagonist DPCPX (100 nM), which reverted CCPA effect on TGF-β1-driven EMT. Different from that observed in response to A2AR activation, PKA inhibition by 5 μM Myr-PKI did not alter CCPA effect, whereas ERK1/2 inhibition by 10 μM PD98059 reverted the strong increase induced by CCPA (Fig. 4b).
At the same time, treatment with TGF-β1 determined morphological changes of MDCK cells. Phalloidin staining showed the presence of elongated F-actin stress fibers when compared to control cells, which exhibited typical epithelial-like morphology. Co-treatment of TGF-β1 and CGS21680 partially reverted fiber rearrangement, whereas CCPA contributed to the disassembly of polymerized actin elicited by TGF-β1 (Fig. 4c).
To evaluate possible involvement of P2Y1 and P2Y11 ATP/ADP receptors (P2Y1R and P2Y11R) in EMT process elicited by TGF-β1, we treated MDCK cells with the selective agonists, MRS2365 or NF546, respectively. When P2Y1R were activated by 30 nM MRS2365, we observed effects similar to those caused by the activation of A1R on the TGF-β1-induced EMT. These effects were reverted by the selective P2Y1 receptor antagonist 1 μM MRS2179. As previously observed, 5 μM Myr-PKI did not modify the effects of MRS2365, whereas 10 μM PD98059 significantly reverted them (Fig. 5a). Therefore, a cross talk between A1R or P2Y1R and TGF-β1 signaling pathways, leading to ERK activation, might be hypothesized.

Effects of P2 adenosine receptor agonists on TGF-β1-induced EMT. a MDCK cells were challenged to 5 ng/mL TGF-β1 for 48 h in the absence or presence of the selective P2Y1R agonist, 30 nM MRS2365, or b the selective P2Y11R agonist, 100 μM NF546. The expression of ZEB1, Slug (left) and Fibronectin, α-SMA (right) was evaluated by RT-PCR analysis. P2Y1R or P2Y11R antagonists, 10 μM MRS2179 or 10 μM NF340, as well as PKA or ERK 1/2 inhibitors, 5 μM Myr-PKI or 10 μM PD98059, were added to culture medium 30 min before TGF-β1 treatment or co-treatment with TGF-β1/P2Y1R or P2Y11R agonists until the end of the experiment. Each column represents the mean ± SEM of at least five independent experiments, and it is expressed as relative amount of mRNA normalized to GAPDH. Student’s t test: *P < 0.05, **P < 0.01, ***P < 0.001 vs. TGF-β1-treated cells; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. TGF-β1/P2Y1R or P2Y11R agonist-treated cells. c Representative photomicrographs of MDCK cells. Cells were challenged with 5 ng/mL TGF-β1 in the absence or presence of 30 nM MRS2365 or 100 μM NF546 for 48 h. The sub-confluent monolayers were stained with rhodamine-phalloidin (red) and nuclei were counterstained with DAPI (blue). Scale bars: 10 μm
Cell exposure to the selective P2Y11R agonist, NF546 (100 μM), significantly decreased the TGF-β1-induced overexpression of ZEB1, Slug, Fibronectin, and α-SMA. These effects, almost completely prevented by cell pre-treatment with the selective antagonist NF340 (10 μM), were partially reduced by the PKA inhibitor, Myr-PKI, or ERK1/2 inhibitor, PD98059 (Fig. 5b). These results were in accordance with multiple signaling pathways linked to activation of Gs/Gq protein coupled P2Y11R.
In agreements with these results, MDCK cells, treated with either TGF-β1 alone or in combination with MRS2365 and stained with phalloidin, exhibited elongation and became larger than untreated cells. F-actins underwent a disassembly and did not appear along the primary axis of the cells but were running in other directions. This was in line with the loss of junction proteins such as E-cadherin, a protein involved in cell-cell interactions in intact renal tubular epithelial cells. When cells were treated with NF546, we observed poor evidence of morphological change compared to control cells (Fig. 5c).
Western blot analyses of Fibronectin and α-SMA were consistent with the above mentioned results of RT-PCR analysis (Fig. 6a–d).

Effects of P1 and P2 purine receptor agonists on TGF-β1-induced EMT. Western blots show the expression of Fibronectin and α-SMA in MDCK cells challenged to 5 ng/mL TGF-β1 for 48 h in the absence or presence of a the selective A2AR agonist, 50 nM CGS21680; b the selective A1R agonist, 50 nM CCPA; c the P2Y1R agonist, 30 nM MRS2365; or d the selective P2Y11R agonist, 100 μM NF546. A2AR, A1R, P2Y1R, or P2Y11R antagonists (50 nM ZM241385, 100 nM DPCPX, 10 μM MRS2179, or 10 μM NF340), as well as PKA or ERK 1/2 inhibitors, 5 μM Myr-PKI or 10 μM PD98059, were added to culture medium 30 min before TGF-β1 treatment or co-treatment with TGF-β1/P1 or P2 receptor agonists until the end of the experiment. Histograms represent data of densitometric analysis, and each column indicates the mean ± SEM of at least three independent experiments and it is expressed as relative protein expression normalized to β-actin. Student’s t test: *P < 0.05, **P < 0.02, ***P < 0.01, § P < 0.001 vs. TGF-β1-treated cells; # P < 0.05, ## P < 0.02, ### P < 0.01, ⌘ P < 0.001 vs. TGF-β1/P1 or P2 receptor agonist-treated cells
The crucial role of P2X7 ATP receptor (P2X7R) in inflammatory processes [35, 36] and tissue fibrosis [37] is well known; furthermore, kidney epithelial P2X7 is involved in inflammation and renal injury [38, 39]. Therefore, we investigated the main signaling pathway involved in P2X7R modulation of EMT in MDCK cells. The exposure of cells to the selective P2X7R agonist BzATP (150 μM) for 48 h did not affect the cell morphology. Since it has been pointed out that in human and murine kidney epithelium [40] and in MDCK cells [41], the expression of P2X7R is relatively low and it may be upregulated by time post-seeding, we prolonged the exposure of MDCK cells to BzATP (150 μM) up to 96 h. At this time point, it was able to induce cell differentiation to myofibroblasts, and 5 ng/mL TGF-β1 added to the culture medium in the last 48 h did not worsen the BzATP-mediated effect (Fig. 7). This result was confirmed by RT-PCR analysis of EMT-related markers, ZEB1, Slug, Fibronectin, and α-SMA. As shown in Table 2, the treatment with 150 μM BzATP for 96 h upregulated the expression of EMT markers to the same extent as for 5 ng/mL TGF-β1 and cell pre-treatment with 150 μM OxATP reverted the agonist effect. As it has been recently demonstrated that BzATP treatment induce the activation of PI3K/Akt and ERK1/2 signaling pathways in prostate cancer cells [42], we exposed cells with ERK1/2 inhibitor, PD98059 (10 μM), added to the culture medium 30 min prior to BzATP treatment until the end of experiment. Intriguingly, PD98059 significantly reduced BzATP-mediated upregulation of all analyzed EMT markers.

Effects of P2X7 receptor agonist on cell morphology. Representative photomicrographs of MDCK cells challenged with 5 ng/mL TGF-β1 for 48 h or P2X7R agonist, 150 μM BzATP, up to 96 h, alone or in the presence of 5 ng/mL TGF-β1, added to the culture medium for the last 48 h of the experiment. The sub-confluent monolayers were stained with rhodamine-phalloidin (red) and nuclei were counterstained with DAPI (blue). Scale bars: 10 μm
Discussion
EMT is a major event in the pathogenesis of fibrosis. Signaling of cAMP is able to counteract interstitial fibrosis by reducing TGF-β1 effect and myofibroblast formation [43]. Specifically, cAMP exerts anti-fibrotic effects by activating both PKA-CREB axis [16] and Epac signaling [18].
It has been reported that cAMP-PKA activation can inhibit the TGF-β1-driven upregulation of collagen type-1 expression in human peritoneal mesothelial cells [44]. Conversely, in breast cancer cells, cAMP seems to promote TGF-β1/Smad3-mediated expression [45]. Furthermore, Weng Lin et al. [46] enlightened the controversial role of cAMP-PKA pathway in MDCK-cell and embryonic kidney-cyst models, where cAMP-PKA showed to exert a protective role by inhibiting TGF-β1-induced ERK1/2 phosphorylation, whereas it exacerbates the fibrotic process in cells not stimulated by TGF-β1.
Based on this evidence, we first individuated the concentration of TGF-β1 able to induce submaximal effects, in order to evaluate pharmacological improvement or worsening of EMT. To this aim, we assayed the time-course and the dose-response curve of TGF-β1 by measuring the expression levels of α-SMA and Fibronectin as well as the transcription factors ZEB1 and Slug, which are commonly considered the most significant markers of myofibroblast differentiation, playing a crucial role in fibrogenesis [47, 48]. Our most suitable experimental conditions have been obtained by using 5 ng/mL of TGF-β1 (almost corresponding to the EC50; see Fig. 1) for 48 h, when the effect of TGF-β1 was already observed. Effectiveness of these parameters has been confirmed by the effects obtained with some cAMP analogues. In our conditions, they showed a biphasic and concentration-dependent modulation of most of EMT-marker expression, which they upregulated or downregulated at lower or higher concentrations, respectively. Therefore, we use concentrations per se unable to affect the expression of the analyzed markers.
We found that cAMP analogues inhibited TGF-β1-induced EMT, either via PKA or Epac pathway. Cell pre-treatment with Myr-PKI, a PKA-selective inhibitor, only in part inhibited the cAMP-mediated anti-fibrotic response, suggesting that the remaining protective activity could be ascribed to the involvement of Epac pathway. Nevertheless, the exposure to the cAMP analogue selective for Epac, 8-CPT-2Me-cAMP, did not show the same efficacy as N6-Ph-cAMP or 8-CPT-cAMP and quite inconsistent results have also been observed for the phosphodiesterase inhibitor, IBMX.
Therefore, our results confirmed the well-known protective activity of the cAMP system on EMT induced by TGF-β1.
Besides the classic Smad mechanisms, more efforts are needed to better elucidate the role of MAPK/ERK cascade [11, 12] in relation to EMT/fibrosis. In order to identify new pharmacological tools for controlling these processes, the effects of agonists of receptors positively linked to cAMP formation as well as antagonists of receptors that, once activated, elicit converging signal to non-Smad pathways of TGF-β1 should be evaluated. Among these receptor systems, a relevant role could be played by purine receptors, as part of the cooperative synergic network called “purinome” [49]. It comprises extracellular purines activating a large number of specific receptors, purine-converting enzymes, either anchored to cell membrane (ecto-enzymes) or released in the intercellular milieu (soluble enzymes) [50,51,52], and membrane carriers for nucleosides and purine bases [53, 54].
Activation of G-protein-coupled P1 adenosine receptors either increases (A2AR and A2BR) or reduces (A1R and A3R) intracellular cAMP levels. Moreover, the stimulation of A1R and A3R can elicit the release of calcium ions from intracellular stores and couple to MAPK pathways, including ERK1/2 and p38 MAPK [55]. Pharmacology studies showed that A2AR modulation could effectively suppress inflammation [56, 57], attenuated collagen deposition in matrix accumulation, and led to attenuation of glomerulonephritis and renal injury [58].
The P2Y subfamily consists of different G protein-coupled receptors (GPCRs) for nucleotides. The Gq-coupled P2Y receptors, P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11, activate phospholipase C pathway that increase intracellular Ca2+. P2Y11 is the only receptor that also couples to Gs and increases intracellular cAMP levels. The remaining P2Y receptors couple to Gi and inhibit adenylyl cyclase activity [59]. P2X receptors are ligand-gated ion channels that play a central role in inflammation, in particular P2X7R [35, 36], whose overexpression has been reported on myeloid and lymphoid cells during several immune and inflammatory processes, including renal injury [38, 39]. Different P2 receptor subtypes are involved in tissue fibrosis [60].
Therefore, in this study, we focused our attention on some purine receptors that play a pivotal role during inflammation and fibrosis [61, 62].
Recently, it has been demonstrated that the activation of some purinergic receptors (P1 and P2) may influence EMT program in several cell types [63], wherein this modulation has been associated with the evolution of several diseases, such as inflammation or cancer.
As expected, A2AR stimulation with CGS21680 exerted protective effects on TGFβ1-induced EMT, as confirmed by the decrease of all mesenchymal markers and transcription factors. Pre-treatment with the A2AR selective antagonist, ZM241385, reverted the agonist-mediated effects. The same result was obtained by using the PKA-selective inhibitor Myr-PKI, thus enlightening the main role of cAMP-PKA-CREB axis in A2AR-mediated effect on EMT. Conversely, no effects were obtained by pre-treating the cells with the inhibitor of ERK1/2 pathway, PD98059. The anti-fibrotic effect of A2AR stimulation was in agreement with that reported in several models of fibrosis [64, 65]. Different from what we observed with A2AR, A1R stimulation with the selective agonist CCPA exacerbated profibrotic effects of TGF-β1. This effect was completely prevented by the pre-treatment with the receptor antagonist DPCPX, whereas it was not affected by the inhibition of PKA pathway. Interestingly, CCPA effect was significantly reduced when the ERK1/2 phosphorylation was inhibited by PD98059. Since the reduction of the TGF-β1-induced marker overexpression mediated by PD98059 was more evident when A1R was activated in addition to TGF-β1 stimulus, it is likely that A1R signaling via β/γ GPCR subunits may cooperate with the MAPK/ERK activated by TGF-β1.
The stimulation of P2Y1R revealed effects similar to those of A1R. Our results provide evidence that P2Y1R signaling exacerbates TGF-β1-induced EMT by causing ERK1/2 phosphorylation but, differently from the A1R, through the increase of intracellular calcium and the PKC activation [59, 66]. A protective effect on the TGF-induced EMT was also caused by stimulation of P2Y11R. This effect was enhanced by ERK1/2 inhibition and was reduced by PKA phosphorylation, thus confirming the simultaneous activation of both Gs and Gq protein subunits coupled to P2Y11R.
Different from all other purine receptor agonists, the P2X7R selective agonist BzATP was able to induce marked changes in cell morphology after a prolonged exposure (96 h), likely due to low amounts of this receptor in MDCK cells [41]. This effect was not worsened by adding TGF-β1 to the culture medium. At the same time, BzATP per se increased the expression of all mesenchymal markers and this effect was reverted by blocking P2X7R, and it was significantly reduced by inhibiting ERK1/2 signaling. These results were in accordance with earlier reports suggesting that P2X7R stimulation elicited the activation of PI3K/Akt and ERK1/2 signaling pathways [42].
Conclusions
In this study, we investigated the interactions between cAMP-PKA and TGF-β1 pathways in EMT. We confirmed the protective role of cAMP stimulation on EMT triggered by TGF-β1. We also provided evidence that the activation of receptor-mediated different pathways, leading to MAPK phosphorylation, determined the overexpression of EMT-related profibrotic markers. Consequently, agonists of purine receptors that stimulate cAMP formation and antagonists of purine receptors that activate MAPK are good candidates for putative anti-fibrotic agents.
Change history
29 September 2023
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1007/s11302-023-09971-w
References
Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression. Nat rev Cancer 2(6):442–454. doi:10.1038/nrc822
Lopez-Novoa JM, Nieto MA (2009) Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol med 1(6–7):303–314. doi:10.1002/emmm.200900043
Hinz B (2010) The myofibroblast: paradigm for a mechanically active cell. J Biomech 43(1):146–155. doi:10.1016/j.jbiomech.2009.09.020
Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176(1):85–97. doi:10.2353/ajpath.2010.090517
Kriz W, Kaissling B, Le Hir M (2011) Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy. J Clin Invest 121(2):468–474. doi:10.1172/JCI44595
Quaggin SE, Kapus A (2011) Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int 80(1):41–50. doi:10.1038/ki.2011.77
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial mesenchymal transition generates cells with properties of stem cells. Cell 133(4):704–715. doi:10.1016/j.cell.2008.03.027
Feng XH, Derynck R (2005) Specificity and versatility in TGF-beta signaling through Smads. Annu rev Cell dev Biol 21:659–693. doi:10.1146/annurev.cellbio.21.022404.142018
Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R (2012) TGF-β-induced activation of mTOR complex 2 drives epithelial mesenchymal transition and cell invasion. J Cell Sci 125(5):1259–1273. doi:10.1242/jcs.095299
Lamouille S, Derynck R (2007) Cell size and invasion in TGF-beta induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 178(3):437–451. doi:10.1083/jcb.200611146
Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE (2008) TRAF6 mediates Smad-independent activation of JNK and p38 by TGF beta. Mol Cell 31(6):918–924. doi:10.1016/j.molcel.2008.09.002
Marchetti A, Colletti M, Cozzolino AM, Steindler C, Lunadei M, Mancone C, Tripodi M (2008) ERK5/MAPK is activated by TGFbeta in hepatocytes and required for the GSK-3beta-mediated snail protein stabilization. Cell Signal 20(11):2113–2118. doi:10.1016/j.cellsig.2008.08.002
Heldin CH, Vanlandewijck M, Moustakas A (2012) Regulation of EMT by TGFb in cancer. FEBS Lett 586(14):1959–1970. doi:10.1016/j.febslet.2012.02.037
Zhang A, Wang MH, Dong Z, Yang T (2006) Prostaglandin E2 is a potent inhibitor of epithelial-to-mesenchymal transition: interaction with hepatocyte growth factor. Am J Physiol Renal Physiol 291(6):F1323–F1331. doi:10.1152/ajprenal.00480.2005
Nieto MA, Huang RY, Jackson RA, Thiery JP (2016) EMT: 2016. Cell 166(1):21–45. doi:10.1016/j.cell.2016.06.028
Schiller M, Dennler S, Anderegg U, Kokot A, Simon JC, Luger TA, Mauviel A, Böhm M (2010) Increased cAMP levels modulate transforming growth factor-beta/Smad-induced expression of extracellular matrix components and other key fibroblast effector functions. J Biol Chem 285(1):409–421. doi:10.1074/jbc.M109.038620
Insel PA, Murray F, Yokoyama U, Romano S, Yun H, Brown L, Snead A, Lu D, Aroonsakool N (2012) cAMP and Epac in the regulation of tissue fibrosis. Br J Pharmacol 166(2):447–456. doi:10.1111/j.1476-5381.2012.01847.x
Cheng X, Ji Z, Tsalkova T, Mei F (2008) Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys sin Shanghai 40(7):651–662 PMC2630796
Breckler M, Berthouze M, Laurent AC, Crozatier B, Morel E, Lezoualc’h F (2011) Rap-linked cAMP signaling Epac proteins: compartmentation, functioning and disease implications. Cell Signal 23(8):1257–1266. doi:10.1016/j.cellsig.2011.03.007
Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, Lee HB (2005) Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J am Soc Nephrol 16(3):667–675. doi:10.1681/ASN.2004050425
Ahmad A, Aboukameel A, Kong D, Wang Z, Sethi S, Chen W, Sarkar FH, Raz A (2011) Phosphoglucose isomerase/autocrine motility factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer res 71(9):3400–3409. doi:10.1158/0008-5472.CAN-10-0965
Zhang A, Jia Z, Guo X, Yang T (2007) Aldosterone induces epithelial-mesenchymal transition via ROS of mitochondrial origin. Am J Physiol Renal Physiol 293(3):F723–F731. doi:10.1152/ajprenal.00480.2006
Noh H, Ha H, Yu MR, Kim YO, Kim JH, Lee HB (2005) Angiotensin II mediates high glucose-induced TGF-beta1 and fibronectin upregulation in HPMC through reactive oxygen species. Perit Dial Int 25(1):38–47
Vyas-Read S, Shaul PW, Yuhanna IS, Willis BC (2007) Nitric oxide attenuates epithelial-mesenchymal transition in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 293(1):L212–L221. doi:10.1152/ajplung.00475.2006
Chan EC, Dusting GJ, Guo N, Peshavariya HM, Taylor CJ, Dilley R, Narumiya S, Jiang F (2010) Prostacyclin receptor suppresses cardiac fibrosis: role of CREB phosphorylation. J Mol Cell Cardiol 49(2):176–185. doi:10.1016/j.yjmcc.2010.04.006
Insel PA, Ostrom RS, Zambon AC, Hughes RJ, Balboa MA, Shehnaz D, Gregorian C, Torres B, Firestein BL, Xing M, Post SR (2001) P2Y receptors of MDCK cells: epithelial cell regulation by extracellular nucleotides. Clin Exp Pharmacol Physiol 28:351–354
Turner CM, King BF, Srai KS, Unwin RJ (2007) Antagonism of endogenous putative P2Y receptors reduces the growth of MDCK-derived cysts cultured in vitro. Am J Physiol Renal Physiol 292:F15–F25
Azroyan A, Cortez-Retamozo V, Bouley R, Liberman R, Ruan YC, Kiselev E, Jacobson KA, Pittet MJ, Brown D, Breton S (2015) Renal intercalated cells sense and mediate inflammation via the P2Y14 receptor. PLoS One. doi:10.1371/journal.pone.0121419
Kempson SA, Edwards JM, Osborn A, Sturek M (2008) Acute inhibition of the betaine transporter by ATP and adenosine in renal MDCK cells. Am J Physiol Renal Physiol 295(1):F108–F117
Saunders C, Keefer JR, Kennedy AP, Wells JN, Limbird LE (1996) Receptors coupled to pertussis toxin-sensitive G-proteins traffic to opposite surfaces in Madin-Darby canine kidney cells. A1 adenosine receptors achieve apical and alpha 2A adrenergic receptors achieve basolateral localization. J Biol Chem 271(2):995–1002
Saunders C, Keefer JR, Bonner CA, Limbird LE (1998) Targeting of G protein-coupled receptors to the basolateral surface of polarized renal epithelial cells involves multiple, non-contiguous structural signals. J Biol Chem 273(37):24196–24206
Zak R (1974) Development and proliferative capacity of cardiac muscle cells. Circ res 35(2):17–26
de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396(6710):474–477. doi:10.1038/24884
Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282(5397):2275–2279
Burnstock G (2016) P2X ion channel receptors and inflammation. Purinergic Signal 12(1):59–67. doi:10.1007/s11302-015-9493-0
Di Virgilio F (2016) P2RX7: a receptor with a split personality in inflammation and cancer. Mol Cell Oncol 3(2):e1010937. doi:10.1080/23723556.2015.1010937
Gentile D, Natale M, Lazzerini PE, Capecchi PL, Laghi-Pasini F (2015) The role of purinergic P2X7 receptors in the inflammation and fibrosis of unilateral ureteral obstruction in mice. Purinergic Signal 11(4):435–440. doi:10.1007/s11302-015-9466-3
Goncalves RG, Gabrich L, Rosario A Jr, Takiya CM, Ferreira MLL, Chiarini LB, Persechini PM, Coutinho-Silva R, Leite M Jr (2006) The role of purinergic P2X7 receptors in the inflammation and fibrosis of unilateral ureteral obstruction in mice. Kidney Int 70(9):1599–1606. doi:10.1038/sj.ki.5001804
Taylor SR, Turner CM, Elliott JI, McDaid J, Hewitt R, Smith J, Pickering MC, Whitehouse DL, Cook HT, Burnstock G, Pusey CD, Unwin RJ, Tam FW (2009) P2X7 deficiency attenuates renal injury in experimental glomerulonephritis. J am Soc Nephrol 20(6):1575–1581. doi:10.1681/ASN.2008060559
Turner CM, Tam FW, Lai PC, Tarzi RM, Burnstock G, Pusey CD, Cook HT, Unwin RJ (2007) Increased expression of the pro-apoptotic ATP-sensitive P2X7 receptor in experimental and human glomerulonephritis. Nephrol Dial Transplant 22(2):386–395. doi:10.1093/ndt/gfl589
Jalilian I, Spildrejorde M, Seavers A, Curtis BL, McArthur JD, Sluyter R (2012) Functional expression of the damage-associated molecular pattern receptor P2X7 on canine kidney epithelial cells. Vet Immunol Immunop 150(3–4):228–233. doi:10.1016/j.vetimm.2012.09.040
Qui Y, Li W, Zhang H, Liu Y, Tian X, Fang W (2014) P2X7 mediates ATP-driven invasiveness in prostate cancer cells. PLoS One 9(12):e114371. doi:10.1371/journal.pone.0114371
Schinner E, Wetzl V, Schlossmann J (2015) Cyclic nucleotide signalling in kidney fibrosis. Int J Mol Sci 16(2):2320–2351. doi:10.3390/ijms16022320
Hung KY, Huang JW, Chen CT, Lee PH, Tsai TJ (2003) Pentoxifylline modulates intracellular signaling of TGF-beta in cultured human peritoneal mesothelial cells: implications for prevention of encapsulating peritoneal sclerosis. Nephrol Dial Transplant 18(4):670–676. doi:10.1093/ndt/gfg141
Oerlecke I, Bauer E, Dittmer A, Leyh B, Dittmer J (2013) Cyclic AMP enhances TGF-beta responses of breast cancer cells by up regulating TGF-beta receptor 1 expression. PLoS One 8(1):e54261. doi:10.1371/journal.pone.0054261
Weng L, Wang W, Su X, Huang Y, Su L, Liu M, Sun Y, Yang B, Zhou H (2015) The effect of cAMP-PKA activation on TGF-beta 1-induced profibrotic signaling. Cell Physiol Biochem 36:1911–1927. doi:10.1159/000430160
Yazhou C, Wenlv S, Weidong Z, Licun W (2004) Clinicopathological significance of stromal myofibroblasts in invasive ductal carcinoma of the breast. Tumor Biol 25(5–6):290–295. doi:10.1159/000081394
Yamashita M, Ogawa T, Zhang X, Hanamura N, Kashikura Y, Takamura M, Yoneda M, Shiraishi T (2012) Role of stromal myofibroblasts in invasive breast cancer: stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 19(2):170–176. doi:10.1007/s12282-010-0234-5
Volontè C, D’Ambrosi N (2009) Membrane compartments and purinergic signalling: the purinome, a complex interplay among ligands, degrading enzymes, receptors and transporters. FEBS j 276(2):318–329. doi:10.1111/j.1742-4658.2008.06793.x
Zimmermann H, Zebisch M, Strater N (2012) Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 8(3):437–502. doi:10.1007/s11302-012-9309-4
Yegutkin GG, Henttinen T, Jalkanen S (2001) Extracellular ATP formation on vascular endothelial cells is mediated by ecto-nucleotide kinase activities via phosphotransfer reactions. FASEB j 15(1):251–260. doi:10.1096/fj.00-0268com
Yegutkin GG, Samburski SS, Jalkanen S (2003) Soluble purine-converting enzymes circulate in human blood and regulate extracellular ATP level via counteracting pyrophosphatase and phosphotransfer reactions. FASEB j 17(10):1328–1330. doi:10.1096/fj.02-1136fje
Novak I (2003) ATP as a signaling molecule: the exocrine focus. News Physiol Sci 18(1):12–17. doi:10.1152/nips.01409.2002
Rose JB, Coe IR (2008) Physiology of nucleoside transporters: back to the future. Physiology (Bethesda) 23:41–48. doi:10.1152/physiol.00036.2007
Antonioli L, Blandizzi C, Pacher P, Haskó G (2013) Immunity, inflammation and cancer: a leading role for adenosine. Nat rev Cancer 13(12):842–857. doi:10.1038/nrc3613
Mazzon E, Esposito E, Impellizzeri D, DI Paola R, Melani A, Bramanti P, Pedata F, Cuzzocrea S (2011) CGS 21680, an agonist of the adenosine (A2A) receptor, reduces progression of murine type II collagen-induced arthritis. J Rheumatol 38(10):2119–2129. doi:10.3899/jrheum.110111
Impellizzeri D, Di Paola R, Esposito E, Mazzon E, Paterniti I, Melani A, Bramanti P, Pedata F, Cuzzocrea S (2011) CGS 21680, an agonist of the adenosine (A2A) receptor, decreases acute lung inflammation. Eur J Pharmacol 668(1–2):305–316
Garcia GE, Truong LD, Chen JF, Johnson RJ, Feng L (2011) Adenosine a(2A) receptor activation prevents progressive kidney fibrosis in a model of immune-associated chronic inflammation. Kidney Int 80(4):378–388. doi:10.1038/ki.2011.101
von Kügelgen I (2006) Pharmacological profiles of cloned mammalian P2Yreceptor subtypes. Pharmacol Ther 110(3):415–432. doi:10.1016/j.pharmthera.2005.08.014
Lu D, Insel PA (2014) Cellular mechanisms of tissue fibrosis. 6. Purinergic signaling and response in fibroblasts and tissue fibrosis. Am J Physiol Cell Physiol 306(9):779–788. doi:10.1152/ajpcell.00381.2013
Burnstock G (2006) Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol rev 58(1):58–86. doi:10.1124/pr.58.1.5
Di Virgilio F (2012) Purines, purinergic receptors, and cancer. Cancer res 72(21):5441–5447. doi:10.1158/0008-5472.CAN-12-1600
Xiao H, Shen HY, Liu W, Xiong RP, Li P, Meng G, Yang N, Chen X, Si LY, Zhou YG (2013) Adenosine A2A receptor: a target for regulating renal interstitial fibrosis in obstructive nephropathy. PLoS One 8(4):e60173. doi:10.1371/journal.pone.0060173
Zhang L, Yang N, Wang S, Huang B, Li F, Tan H, Liang Y, Chen M, Li Y, Yu X (2011) Adenosine 2A receptor is protective against renal injury in MRL/lpr mice. Lupus 20:667–677. doi:10.1177/0961203310393262
Huang X, He Y, Chen Y, Wu P, Gui D, Cai H, Chen A, Chen M, Dai C, Yao D, Wang L (2016) Baicalin attenuates bleomycin-induced pulmonary fibrosis via adenosine A2a receptor related TGF-β1-induced ERK1/2 signaling pathway. BMC Pulm 16(1):132. doi:10.1186/s12890-016-0294-1
Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol rev 50(3):413–492
Acknowledgements
This study was partially supported by funds to P. Di Iorio from the Italian Ministry of Education, University and Research (MIUR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
Mariachiara Zuccarini declares that she has no conflict of interest.
Patricia Giuliani declares that she has no conflict of interest.
Silvana Buccella declares that she has no conflict of interest.
Valentina Di Liberto declares that she has no conflict of interest.
Giuseppa Mudò declares that she has no conflict of interest.
Natale Belluardo declares that she has no conflict of interest.
Marzia Carluccio declares that she has no conflict of interest.
Margherita Rossini declares that she has no conflict of interest.
Daniele Filippo Condorelli declares that he has no conflict of interest.
Michel Piers Rathbone declares that she has no conflict of interest.
Francesco Caciagli declares that he has no conflict of interest.
Renata Ciccarelli declares that she has no conflict of interest.
Patrizia Di Iorio declares that she has no conflict of interest.
Ethical approval
This article does not contain any studies with participants or animals performed by any of the authors.
Additional information
Mariachiara Zuccarini and Patricia Giuliani are co-first authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zuccarini, M., Giuliani, P., Buccella, S. et al. RETRACTED ARTICLE: Modulation of the TGF-β1-induced epithelial to mesenchymal transition (EMT) mediated by P1 and P2 purine receptors in MDCK cells. Purinergic Signalling 13, 429–442 (2017). https://doi.org/10.1007/s11302-017-9571-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-017-9571-6