
Abstract
In the last 5 years, many efforts have been conducted searching potent and selective human A3 adenosine antagonists. In this field several different classes of compounds, possessing very good affinity (nM range) and with a broad range of selectivity, have been proposed. Recently, our group synthesized a new series of pyrazolo-triazolo-pyrimidines bearing different substitutions at the N5 and N8 positions, which have been described as highly potent and selective human A3 adenosine receptor antagonists. The present review summarizes available data and provides an overview of the structure–activity relationships found for this class of human A3 adenosine receptor antagonists.
Similar content being viewed by others
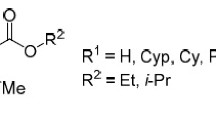

Avoid common mistakes on your manuscript.
Introduction
In the last 10 years the pyrazolo-triazolo-pyrimidine nucleus (1) represented an attractive key intermediate for obtaining adenosine receptor antagonists due to its strong structural correlation with the nonselective adenosine receptor antagonist CGS15943 (2) [1, 2] (Figure 1).

Structural similarities between pyrazolo-triazolo-pyrimidines and reference compound CGS15943 [1]
The great advantage of this nucleus with respect to the reference compound (2) is related to a large number of substitutions that could be done on this nucleus, such as in positions N7, N8, N5, C9, or C2 opening kaleidoscopic possibilities for different substituted heterocycles as adenosine receptor antagonists. The aim of this review was to briefly summarize the great efforts made on this class of compounds, resulting in a complete structure–activity relationship (SAR) profile of the pyrazolo-triazolo-pyrimidine family.
Substitutions at the N7 and N8 positions
The first example of adenosine receptor antagonists with the pyrazolo-triazolo-pyrimidine nucleus was reported by Gatta and coworkers [3], who proposed a compound named 8FB-PTP (3), which displayed potent binding to but no selectivity versus A2A adenosine receptors (Figure 2).

Taking into account this experimental observation and considering the limited number of compounds reported by Gatta et al., our group decided to extensively investigate the effect of substitutions on the pyrazole moiety. From earlier studies, the importance of the free amino group at 5-position and the effect of the substituent on the pyrazole ring seems to be fundamental for both high affinity and selectivity for the A2A adenosine receptor subtype. In particular, substitutions at 7-position improve the selectivity for the A2A adenosine receptor whereas the same substitutions at 8-position increase the potency both at the A1 and A2A receptors with low levels of selectivity as a consequence, as indicated for the N7-n-butyl (4) and the N8-n-butyl (5) derivatives [4, 5] (Figure 2).
An accurate investigation of the effect of the chain at the N7 position clearly demonstrated that the presence of an aralkyl chain seems to be essential for both potency and selectivity at the A2A adenosine receptors. In fact, two selected compounds, named SCH 58261 (6) and SCH 63390 (7) proved to be the most potent and selective A2A adenosine receptor antagonists ever reported, both in rat and human models [5] (Figure 3).

Nevertheless, this class of compounds presents a significant problem in relation to the poor water solubility, which compromises the use of this compound as pharmacological tool. To overcome this problem, several polar moieties on the side chain of the pyrazole nucleus have been introduced. In particular, the introduction of a hydroxyl function at the para position of the phenyl ring of compounds (6) and (7), as in one of the most potent and selective A2A antagonist ZM241385 (4-[2-[[7-amino-2-(2-furyl) [1,2,4]-triazolo[2,3-a] [1,3,5]triazin-5-yl]amino]ethyl]phenol) [6a], led to derivatives (8) (5-amino-7-[β-(4-hydroxyphenyl)ethyl]-2-(2-furyl)pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine) and (9) (5-amino-7-[3-(4-hydroxyphenyl)propyl]-2-(2-furyl)pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine), which not only showed a better hydrophilic character but also a significant increase of both affinity and selectivity for the A2A adenosine receptor subtype, suggesting that most probably, a hydrogen bond is involved in the receptor recognition via this part of the ligand [6] (Figure 4).

Hydrophilic pyrazolo-triazolo-pyrimidines as A2A adenosine receptor antagonists [6b]
To understand the nature of such a hypothetical hydrogen bond, compound SCH 442416 (10) was synthesized. This derivative showed even higher affinity and selectivity for the A2A adenosine receptor, which makes it a candidate as a tool for positron emission tomography (PET) studies in its 11C-labeled form [7]. In addition, this derivative confirms the role of a hydrogen bond via the pyrazolo side chain.

However, the introduction of oxygenated groups could not be considered sufficient to confer water solubility. For this reason, carboxylic (11) and sulfonic (12) moieties were introduced, which contributed highly to the water solubility, in particular, the sulfonic moiety. However, in some cases, a loss of affinity with respect to reference compounds (8, 9) for the A2A adenosine receptor was the consequence. In contrast, the introduction of an amino group at para position of the phenyl ring in the side chain (13) gave better results in terms of affinity and selectivity for the A2A adenosine receptor subtype, but unfortunately, the water solubility was not optimal for a possible therapeutic use. From these advanced studies, it seemed very hard to find a useful combination of affinity, selectivity, and acceptable characteristics of bioavailability [8] (Figure 5).

Structures and binding affinities of different water-soluble pyrazolo-triazolo-pyrimidines [8]
The binding data for these compounds revealed another relevant aspect relating to the N7 and N8 pattern of substitutions (e.g., compounds 6 and 14) (see also Figure 3) [5]. It is quite evident that the N7 derivative (6) is totally inactive at the human A2B and A3 receptors whereas the N8 isomer (14) showed a slight affinity profile for these two receptor subtypes.

This experimental observation strongly suggests a fundamental role of space occupancy of the substitutions on the pyrazole nucleus for recognition of the different receptor subtypes, and it represented a staring point for further studies.
Substitution at the N5 position
These results described above led us to thoroughly investigate this structure as a possible template for other subtype selective ligands, in particular, versus the human A3 and A2B adenosine receptors. Taking into account these data and the fact that MRS 1220, (15) [9] was the most potent but not highly selectivte A3 adenosine receptor antagonist reported in the literature, and considering the structural relation to CGS15943 (1) and the pyrazolo-triazolo-pyrimidine core, we modified the N5 position of this nucleus in an attempt to obtain antagonists for the human A3 adenosine receptor.

The planned modification was based on the introduction of a phenylcarbamoyl chain (4-OMe or 3-Cl substituted) present in a previously reported series of N6 (substituted phenylcarbamoyl) adenosine-5’-uronamides, as A3 agonists, of general formula (16), [4, 5], at the N5 position of the pyrazolo-triazolo-pyrimidine scaffold [10, 11] (Figure 6).

Rational design of the human A3 adenosine receptor antagonists
A preliminary study led to the discovery of compound (5-[[(4-methoxyphenyl)amino]carbonyl]amino-8-ethyl-2-(2-furyl)-pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine (17), which turned out to be the most potent and selective human A3 adenosine receptor antagonist ever reported and yet was totally inactive in a rat model [12].

This result represented the starting point for an intensive investigation of this new class of human A3 adenosine receptor antagonist. A classic SAR approach for studying this new class of human A3 adenosine receptor antagonists was performed. We found an interesting correlation between the calculated molecular volume of pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives and their experimental Ki values. In fact, it was observed that the A3 affinities decrease with increasing molecular volume values at the N8 position. All these observations suggested that small alkyl groups at the N8 pyrazole nitrogen combined with the N5-(4-methoxy)phenylcarbamoyl substitution afford the best compounds in terms of affinity and selectivity at the human A3 adenosine receptors [13].

In particular, when the N8-methyl and N5-(4-methoxy)phenylcarbamoyl substitutions were combined, the most potent and selective human A3 adenosine antagonist (18) was obtained [13]. A molecular modeling investigation, reported in the next session/paragraph, was derived in order to rationalize these experimental results. This study also permitted the introduction of an allyl chain at the N8 position that, after reduction with tritium, afforded a labeled form named MRE3008-F20 (19), which was the first antagonist radioligand utilized for studying the human A3 adenosine receptor, showing a KD value of 0.82 nM and a Bmax of 297 fmol/mg protein (Figure 7) [14, 15].

As for the A2A antagonists, these derivatives also showed very low water solubility, which limits their use for therapeutic purpose. Considering that only the phenyl ring is a suitable core for the introduction of polar substituents, we investigated which position (ortho, meta, para) and substituent (e.g., Br, SO3H, F, H, CH3, CF3) could be suitable to solve this problem while maintaining a small substituents (methyl) at the N8 position. The molecular modeling study performed on the synthesized compounds suggested that the steric factors at the ortho or para position on the phenyl ring play a fundamental role for affinity at the human A3 receptors, and a very rigid steric control is present at the meta position. In fact, compound (20) was found to be the best compound of the series whereas derivative (21), which is completely water soluble, showed a dramatic loss of potency and selectivity [16] (Figure 8).

Structures and binding affinities of the most interesting compounds modified at the phenyl ring [16]
Starting from experimental observation that bulky substituents are not well tolerated on the phenyl ring (see compounds 20 and 21), the bioisosteric replacement of the phenyl ring with the 4-pyridyl moiety was performed, thus providing higher water solubility while avoiding the steric hindrance of a substituent at the para position, which seemed to be responsible for the reduction of human A3 adenosine receptor affinity. This approach led us to the discovery of the most potent (pM range), selective (>10,000)—and most importantly—water-soluble (15 mM) (22) human A3 adenosine receptor antagonist.

The high potency of this compound was confirmed by functional assay. An analysis of the antagonism of agonist-induced inhibition of cyclic adenosine monophosphate (cAMP) production in Chinese hamster ovary (CHO) cells expressing the human A3 receptor indicated a KB value of 0.20 nM for compound (22) [17]. In this case, the support of molecular modeling was fundamental not only for the design of this derivative but also for understanding why the presence of an endocyclic nitrogen was capable of inducing a significant increase of potency in binding interaction. In fact, strong electrostatic interactions appeared to occur between the positively charged pyridinium moiety of (22) and the carbonyl oxygen atoms of Asn274 (N+H⋯ OC distance = 2.5 Å) and Asn278 (N+H⋯ OC distance = 3.1 Å), both located on transmembrane (TM)7, which could be considered responsible for the increase in affinity
A quite similar approach was utilized for the improvement of the affinity at human A2B adenosine receptors. In fact, CGS15943 not only binds to human A3 receptors but displays good affinity for the A2B subtype. Jacobson and coworkers observed that the introduction of polar moieties, such as γ-aminobutyryl amide (23), results in increased potency at the A2B adenosine receptors but a complete lack of selectivity versus the A1 and A2A subtypes. However, the presence of apolar chains, such as the N5-pyvaloyl (24) or the N5-tert-butyloxycarbony derivative (25), displayed less potency than derivative (23) at the A2B adenosine receptors but an increased selectivity versus the other receptor subtypes, indicating a preliminary SAR profile of this class of compounds as A2B adenosine receptor antagonists (Figure 9) [18].

Structures and binding affinities of triazolo-quinazolines as A2B adenosine receptor antagonists [18]
Considering these data, we performed a similar approach on the pyrazolo-triazolo-pyrimidines. We observed that the N5 unsubstituted derivatives (14, 26) possess high affinity at the human A2B adenosine receptors but a complete lack of selectivity. Instead, its substitution with a γ-aminobutyryl amide (27) produces a decrease of affinity at the A2B adenosine receptors but was found to be slightly selective versus the A2A subtype [19] (Figure 10).

Structures and binding affinities of pyrazolo-triazolo-pyrimidines as A2B adenosine receptor antagonists [19]
An improvement of this class of compounds was achieved by an optimized pattern of substitutions at the N5 and N8 positions. In fact, in parallel studies on human A3 adenosine antagonists, we observed that replacement of the phenylcarbamoyl moiety at the N5 position with a phenylacetyl group (e.g., compound 28) produces a decrease in affinity at the human A3 adenosine receptor affinity and a retention or improvement versus the A2B subtype. In fact, a combination of a naphthyl acetyl moiety at the N5 position and a phenylpropyl group (characteristic of A2A antagonists) at the N8 position led to compound (29), which was found to be quite potent and selective for the A2B adenosine receptors [20] (Figure 11).

Structures and binding affinities of N5 arylacetyl pyrazolo-triazolo-pyrimidines as A2B adenosine receptor antagonists [20]
These results seem to suggest that bulky substituents at both the N5 and N8 positions could lead to potent and selective A2B adenosine receptor antagonists [20]. A theoretical comparison of the putative TM binding motif of compound (29) on both human A2B and human A3 receptors was able to elucidate these results, explaining why such small modifications could significantly change the biological profile of this class of compounds.
Molecular modeling
We recently developed a combined target-based and ligand-based drug design approach to define a pharmacophore model of human A3 receptor antagonists, in order to lead the discovery and the structural refinement of new potent and selective human A3 receptor antagonists. In particular, we developed a rhodopsin-based model of the human A3 receptor to provide more accurate information about the putative binding site of the pyrazolo-triazolo-pyrimidines [16, 17, 20–24]. As already reported, this binding site for A3 agonists and antagonists was located in the upper region of the TM helical bundle of the receptor. The human A3 receptor model revealed a central pocket surrounded by helices 1–7 and 5–6, as TM4 is not part of the cavity wall. TMs 3, 5, 6, and 7 seem to be crucial for the recognition of A3 receptor ligands, and particular care has been given to the second extracellular loop (EL2). EL2, as for rhodopsin, folds back over TM helices, limiting the size of the ligand recognition site and preventing access to the cavity from the periplasm [25]. Docking studies, validated by site-directed mutagenesis analysis, pointed out several residues in transmembrane domains 3 and 5 and in the EL2 that seem to be critical for ligand recognition, including His95, Trp243, Ser247, Asn250, and Lys152 [23, 24, 26]. The molecular docking study was carried out on more than 200 pyrazolo-triazolo-pyrimidine derivatives to recognize the hypothetical binding motif of human A3 receptor antagonists and to define a target-based pharmacophore model. Consequently, to generate a three-dimensional (3D)-driven pharmacophore hypothesis, the docked low energy conformations of pyrazole-triazolo-pyrimidine were used as a structural template. Therefore, we identified the hypothetical binding site of these derivatives and proposed a “Y-shaped” pharmacophore model [22]. Interestingly, all derivatives share a common binding motif inside the TM region of human A3 receptors: the pyrazole-triazolo-pyrimidine moiety is surrounded by TMs 3, 5, 6, and 7, with the furan ring and the N8 substituents pointing toward the EL2 and the carbamoyl moiety in the 5-position oriented toward the intracellular environment. The Y-shape binding motif is defined by a peculiar and highly conserved binding mode: the furan ring is positioned between TM5 and TM3 whereas the N8 substituents are surrounded by TM2 and TM7.
Moreover, analyzing in detail the 3D pharmacophore model, we identified five regions that seem to be crucial for antagonists recognition. In particular, all pyrazole-triazolo-pyrimidine derivatives present the carbamoyl moiety in the 5-position such that it is surrounded by two polar amino acids: His95 (TM3) and Ser247 (TM6). The major structural difference between the hypothetical binding sites in adenosine receptor subtypes is that the A3 receptor does not contain the histidine residue in TM6 common to all A1 (His251 in hA1) and A2 (His250 in hA2A) receptors. This histidine has been shown to participate in both agonist and antagonist binding to A2A receptors whereas in the A3 receptor, the histidine is replaced with a serine residue (Ser247 in hA3) [27, 28]. The polar amino acids stabilize interactions with the carbamoyl moiety, and consequently, the carbamoyl phenyl ring is oriented in the middle of the TM bundle (Figure 12).

General topology of the human A3 receptor model. Reference compound 20 (colored in magenta) is docking inside the transmembrane recognition site. Side chains of some amino acids important for ligand recognition are highlighted
In particular, 2.6 Å separate the N-H of His95 (TM3) and the oxygen atom of the carbamoyl group, appropriately oriented to form a H-bonding interaction. The side chain of Ser247 (TM6) is within hydrogen-bonding distance of NH of the carbamoyl group at 2.9 Å. The phenyl ring of the carbamoyl moiety takes place in a receptor region that is hydrophobic and characterized by three nonpolar amino acids: Ile98 (TM3), Ile186 (TM5), and Leu244 (TM6). As a consequence of this, polar substituents, such as a sulfonic acid group at the para position of the phenyl ring, are not very well tolerated [16]. Moreover, around this para position, it seems that a steric control is present because of the very limited empty space between TM5 and TM6. In fact, there is a reduction in the affinity at the human A3 adenosine receptors of the N5 phenyl ring derivatives substituted at the para position [16, 17]. In the case of meta substitutions, we observed a similar steric control when the meta residues are larger than hydrogen: a significant affinity reduction at the human A3 receptor is due to the strong steric repulsion among the meta substituents and amino acid side chains of TM6 and TM7. In contrast, the residues at the ortho position seem to occupy an empty region of the binding cavity.
Another important pharmacophore feature is located in a highly conserved region delimited by Phe168 (EL2) and Phe182 (TM5). There is a probable π–π interaction between the furan ring of the pyrazole-triazolo-pyrimidine and these two phenylalanine residues. In particular, the amino acids corresponding to Leu90 (TM3) and Phe182 in the human A2A receptor was found to be essential for the binding of both agonists and antagonists [28]. Moreover, the nonpolar amino acid Leu90, together with Leu246 (TM6) and Ile268 (TM7), delimited a peculiar hydrophobic pocket where all the substituents at the N8 position are located, with a good tolerance for the hydrophobic ones. However, in the presence of appropriate substituents, hydrogen bonding can be established with two hydrophilic amino acids, namely, Ser175 (EL2) and the His272 (TM7), located on the border of this hydrophobic pocket. [13] However, other aromatic rings, such as phenyl or thiophenyl, seem to be less well tolerated by the receptor-binding pocket, probably due to their less favorable steric complementaries [29].
Another strong hydrogen bonding interaction occurs between Asn250 (TM6) and the triazolo ring of the pyrazole-triazolo-pyrimidine moiety. The N-H of Asp250 and the N4 of the triazolo ring group are, in fact, separated by 2.5 Å and appropriately oriented to form an H-bonding interaction. The two polar amino acids Thr94 (TM3) and Ser97 (TM3) were shown to be important for the coordination of agonist but not for antagonist binding at A1 and A2A receptors, respectively. Therefore, no direct interactions have been predicted between them and the antagonist structure.
In parallel with the docking studies and the target-based pharmacophore model generation, we derived a comparative molecular field analysis (CoMFA) on the pyrazole-triazolo-pyrimidine analogue binding data, as alternative scoring function for the prediction of ligand-receptor-binding affinity [22, 24]. The analysis was performed considering the previous results of the docking approach. The CoMFA approach consists of the computation of steric and electrostatic field variables calculated at grid points surrounding the whole molecule, followed by a partial least squares (PLS) on these 3D structure descriptors and on the biological activity. The PLS analysis, followed by cross-validation, showed very good values of correlation, meaning high predictivity power of the quantitative structure activity relationship (QSAR) model. The CoMFA results can be observed and analyzed as a contour plot and regression plots. The contour plot consists of colored polyhedra that describe the regions of space where the steric and electrostatic fields are predicted to have an effect on binding affinity. The yellow and the blue contours correspond to regions of the field where increase steric bulk and electronegative character is associated with a decrease of receptor affinity. In particular, we observed that the contour map displays a green polyhedron nearby the N5-carbamoyl moiety, suggesting enhanced A3 receptor affinity if we introduce sterically bulky substituents in this position. As already described, this steric-bulk-favorable region nicely fits with the receptor region around the phenyl ring of the carbamoyl moiety, characterized by the three nonpolar amino acids Ile98(TM3), Ile187(TM5), and Leu244(TM6). On the other side, we observed two red polyhedra located around to N8 substituents and to the N5-carbamoyl moiety, complementary to the polar amino acids Ser175 (EL2), His95 (TM3), Ser247 (TM6), and His272 (TM7) present in this region in the A3-receptor-binding-site model. Similarly, the blue polyhedron close to the N5 phenyl ring is complementary to the nonpolar residues Ile98 (TM3), Ile186 (TM5), Phe239 (TM6), and Leu244 (TM6) (Figure 13).

Side view of the 20-A3 complex model. The side chains of some crucial important residues in proximity (≤5 Å) to the docked pyrazolo-triazolo-pyrimidine molecule are highlighted and labeled: Leu90 (TM3), His 95 (TM3); Phe182 (TM5), Ile186 (TM5); Trp243 (TM6); Ser247 (TM6), Asn250 (TM6), Ser271 (TM7), His272 (TM7), Ser275 (TM7). The steric and the electrostatic contour plots, obtained from the comparative molecular field analysis (CoMFA), are included into ligand binding cavity. The yellow and the blue polyhedra correspond to regions of the field that are predicted to decrease the A3 receptor affinity whereas the green and the red regions are predicted to increase binding affinity
In Figure 13, we present the CoMFA contour plot of a pyrazole-triazolo-pyrimidine derivative docked inside the human A3-receptor-binding-site model. We can see that all steric and electrostatic features are coherent with the SAR previously described in the molecular docking analysis. This combined approach between a very consolidated target-based approach, such as molecular docking, and a solid quantitative ligand-based methodology, such as CoMFA, is very advantageous for the identification of common structural features of pyrazolo-triazolo-pyrimidines important for the affinity at the A3 receptor. We have, in fact, had really good results in validation of this combined approach, predicting with high precision the binding affinity values of new derivatives with a different spectrum of affinity at the human A3 receptor.[22, 24]
Substitutions at the C2 and C9 positions
Very recently, a number of pyrazolo-triazolo-pyrimidines with classic substituents at N7 and N5 positions have been modified at the C2 or C9 positions. In general, it has been observed that the introduction of a substituent on the C9 position (e.g., SCH3, NHCH2CH3) led to retention of a receptor affinity but a complete loss of selectivity between different receptor subtypes. Indeed, substitution of the furan ring with phenyl or alkoxyphenyl rings led to a loss of affinity at A2A, A2B, and A3 adenosine receptors only. At the A1 receptor, a high nanomolar affinity is still present. Also, the introduction of polar substituents (by Mannich reaction) on the furan ring led to complete inactivity. Som examples (30, 31) are depicted in Figure 14.

Structures and binding profile of C2 and C9 substituted pyrazolo-triazolo-pyrimidines [30]
These data clearly indicate that position 9 plays a role for selectivity whereas the presence of an unsubstituted furan ring at the C2 position plays a fundamental role in ligand-receptor recognition [30].
Concluding remarks
All the work performed on the pyrazolo-triazolo-pyrimidine nucleus resulted in potent and selective ligands for A2A, A2B, and A3 adenosine receptor subtypes whereas for the A1 subtype, potency was achieved but selectivity was very poor. All these experimental observations resulted in a quite significant SAR profile of this nucleus for the adenosine receptor subtypes. In addition, in the case of A3 adenosine receptor, all these studies allowed development of a receptor-based pharmacophore model with high levels of predictivity for newly designed compounds, as recently reported.
Abbreviations
- CGS15943:
-
(9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine)
- 8FB-PTP:
-
5-amino-8-(4-fluorobenzyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine)
- SCH 58261:
-
5-amino-7-(β-phenylethyl)-2-(2-furyl)pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine
- SCH 63390:
-
5-amino-7-(3-phenylpropyl)-2-(2-furyl)pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine
- SCH 442416:
-
5-amino-7-[3-(4-methoxyphenyl)propyl]-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine
- PET:
-
positron emission tomography
- MRS 1220:
-
N-[9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-yl]benzene-acetamide
- MRE3008-F20:
-
5-[[(4-Methoxyphenyl)amino]carbonyl]amino-8-propyl-2-(2-furyl)-pyrazolo[4,3-e] 1,2,4-triazolo[1,5-c]pyrimidine
- TM:
-
transmembrane domain
References
Baraldi PG, Cacciari B, Romagnoli R, Merighi S, Varani K, Borea PA, Spalluto G (2000) A3 Adenosine receptor ligands; history and perspectives. Med Res Rev 20:103–128
Baraldi PG, Cacciari B, Borea PA, Varani K, Pastorin G, Da Ros T, Tabrizi MA, Fruttarolo F, Spalluto G (2002) Pyrazolo-triazolo-pyrimidines as adenosine receptor antagonists: A possible template for adenosine receptor subtypes? Curr Pharm Des 8:2299–2332
(a) Gatta F, Del Giudice MR, Borioni A, Borea PA, Dionisotti S, Ongini E (1993) Synthesis of imidazo[1,2-c]pyrazolo[4,3-e] pyrimidines, pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidines and triazolo[5,1-i]purines: new potent A2 adenosine receptor antagonists. antagonists. Eur J Med Chem 28:569–576; (b) Dionisotti S, Conti A, Sandoli D, Zocchi C, Gatta F, Ongini E (1994) Effects of the new A2 adenosine receptor antagonist 8FB-PTP, an 8 substituted pyrazolo-triazolo-pyrimidine, on in vitro functional models. Br J Pharmacol 112: 659–665
Baraldi PG, Manfredini S, Simoni D, Zappaterra L, Zocchi C, Dionisotti S, Ongini E (1994) Synthesis of new pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine and 1,2,3-triazolo[1,5-c]pyrimidine displaying potent and selective activity as A2A adenosine receptor antagonists. Bioorg Med Chem Lett 4:2539–2544
Baraldi PG, Cacciari B, Spalluto G, Pineda de las Infantas y Villatoro MJ, Zocchi C, Dionisotti S, Ongini E (1996) Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives: potent and selective A2A adenosine antagonists. J Med Chem 39:1164–1171
(a) Poucher SM, Keddie JR, Singh P, Stoggall SM, Caulkett PWR, Jones G, Collis MG (1995) The in vitro pharmacology of ZM 241385, a potent, nonxanthine, A2A selective adenosine receptor antagonist. Br J Pharmacol 115:1096–1102; (b) Baraldi PG, Cacciari B, Spalluto G, Bergonzoni M, Dionisotti S, Ongini E, Varani K, Borea PA (1998) Design, synthesis, and biological evaluation of a second generation of pyrazolo[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidines as potent and selective A2A adenosine receptor antagonists. J Med Chem 41:2126–2133
Todde S, Moresco RM, Simonelli P, Baraldi PG, Cacciari B, Spalluto G, Varani K, Monopoli A, Matarrese M, Carpinelli A, Magni F, Galli Kienle M, Fazio F (2000) Design, radiosynthesis, and biodistribution, of a new potent and selective ligand for in vivo imaging of the adenosine A2A receptor system using positron emission tomography. J Med Chem 43:4359–4362
Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Monopoli A, Ongini E, Varani K, Borea PA (2002) 7-Substituted 5-amino-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines as A2A adenosine receptor antagonists: a study on the importance of modifications at the side chain on the activity and solubility. J Med Chem 45:115–126
Kim YC, Ji XD, Jacobson KA (1996) Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J Med Chem 39:4142–4148
Baraldi PG, Cacciari B, Spalluto G, Ji XD, Olah ME, Stiles G, Dionisotti S, Zocchi C, Ongini E, Jacobson KA (1996) Novel N6-(substituted-phenylcarbamoyl) adenosine-5’-uronamides as potent agonists for A3 adenosine receptors. J Med Chem 39:802–806
Baraldi PG, Cacciari B, Pineda de Las Infantas MJ, Romagnoli R, Spalluto G, Volpini R, Costanzi S, Vittori S, Cristalli G, Melman N, Park KS, Ji XD, Jacobson KA (1998) Synthesis and biological activity of a new series of N6-arylcarbamoyl, 2-(Ar)alkynyl-N6-arylcarbamoyl, and N6-carboxamido derivatives of adenosine-5’-N-ethyluronamide as A1 and A3 adenosine receptor antagonists. J Med Chem 41:3174–3185
Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Klotz KN, Leung E, Varani K, Gessi S, Merighi S, Borea PA (1999) Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A3 adenosine receptor antagonists. J Med Chem 42:4473–4478
Baraldi PG, Cacciari B, Romagnoli R, Spalluto G, Moro S, Klotz KN, Leung E, Varani K, Gessi S, Merighi S, Borea PA (2000) Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A3 adenosine receptor antagonists: Influence of the chain at N8 pyrazole nitrogen. J Med Chem 43:4768–4780
Baraldi PG, Cacciari B, Romagnoli R, Varani K, Merighi S, Gessi S, Borea PA, Leung E, Hickey SL, Spalluto G (2000) Synthesis and preliminary biological evaluatuion of [3H]MRE3008-F20: the first high affinity radioligand antagonist for the human A3 adenosine receptors. Bioorg Med Chem Lett 10:209–211
Varani K, Merighi S, Gessi S, Klotz KN, Leung E, Baraldi PG, Cacciari B, Spalluto G, Borea PA (2000) [3H]MRE3008-F20: a novel antagonist radioligand for the pharmacological and biochemical characterization of human A3 adenosine receptors. Mol Pharmacol 57:968–975
Baraldi PG, Cacciari B, Moro S, Spalluto G, Pastorin G, Da Ros T, Klotz KN, Varani K, Gessi S, Borea PA (2002) Synthesis, biological activity, and molecular modeling investigation of new pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as human A3 adenosine receptor antagonists. J Med Chem 45:770–780
Maconi A, Pastorin G, Da Ros T, Spalluto G, Gao ZG, Jacobson KA, Baraldi PG, Cacciari B, Varani K, Borea PA (2002) Synthesis, Biological properties and molecular modeling investigation of the first potent, selective and water soluble human A3 adenosine receptor antagonist. J Med Chem 45:3579–3582
Kim YC, de Zwart M, Chang L, Moro S, Jacobien K, Frijtag DK, Melman N, IJzerman AP, Jacobson KA (1998) Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) having high potency at the human A2B and A3 receptor subtypes. J Med Chem 41:2835–2845
Baraldi PG, Cacciari B, Romagnoli R, Klotz KN, Spalluto G, Varani K, Gessi S, Merighi S, Borea PA (2001) Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as adenosine receptor ligands: A starting point for searching A2B adenosine receptor antagonists. Drug Dev Res 53:225–235
Pastorin G, Da Ros T, Spalluto G, Deflorian F, Moro S, Cacciari B, Baraldi PG, Gessi S, Varani K, Borea PA (2003) Pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives as adenosine receptor antagonists. Influence of the N5 substituent on the affinity at the human A3 and A2B adenosine receptor subtypes: a molecular modeling investigation. J Med Chem 46:4287–4296
Moro S, Deflorian F, Spalluto G, Pastorin G, Cacciari B, Kim SK, Jacobson KA (2003) Demystifying the three-dimensional structure of G preotein-coupled receptors (GPCRs) with the aid of molecular modeling. Chem Commun (Cambridge) 21(24):2949–2956
Moro S, Braiuca P, Deflorian F, Ferrari C, Pastorin G, Cacciari B, Baraldi PG, Varani K, Borea PA, Spalluto G (2005) Combined target-based and ligand-based drug design approach as tool to define a novel 3D-pharmacophore model of human A3 adenosine receptor antagonists: pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as a key study. J Med Chem 48:152–162
Moro S, Bacilieri M, Deflorian F, Spalluto G (2006) G protein-coupled receptors as challenging druggable targets: insights from in silico studies. New J Chem 30:301–308
Moro S, Bacilieri M, Deflorian F, Spalluto G (2006) Ligand-based homology modeling as attractive tool to inspect GPCR structural plasticity. Curr Pharm Des 12:2175–2185
Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M (2000) Crystal structure of rhodopsine: a G protein couplet receptor. Science 289:739–745
Moro S, Deflorian F, Bacilieri M, Spalluto G (2006) Novel strategy for the design of new potent and selective human A3 receptor antagonists: an update. Curr Med Chem 13:639–645
Gao ZG, Chen A, Barak D, Kim SK, Muller CE, Jacobson KA (2002) Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J Biol Chem 277:19056–19063
Jiang Q, Lee BX, Glashofer M, van Rhee AM, Jacobson KA (1997) Mutagenesis reveals structure-activity parallels between human A2A adenosine receptors and biogenic amine G protein-coupled receptors. J Med Chem 40:2588–2595
Pastorin G, Da Ros T, Bolcato C, Montopoli C, Moro S, Cacciari B, Baraldi PG, Varani K, Borea PA, Spalluto G (2006) Synthesis and biological studies of a new series of 5-heteroarylcarbamoylamino-pyrazolo[4,3-e]1,2,4triazolo[1,5-c]pyrimidines as human A3 adenosine receptor antagonists. Influence of the heteroaryl substituent on binding affinity and molecular modeling investigation. J Med Chem 49:1720–1729
Baraldi PG, Fruttarolo F, Tabrizi MA, Preti D, Romagnoli R, El-Kashef H, Moorman A, Varani K, Gessi S, Merighi S, Borea PA (2003) Design, synthesis and biological evaluation of C9 and C2 substituted pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines as new A2A and A3 adenosine receptor antagonists. J Med Chem 46:1229–1241
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cacciari, B., Bolcato, C., Spalluto, G. et al. Pyrazolo-triazolo-pyrimidines as adenosine receptor antagonists: A complete structure–activity profile. Purinergic Signalling 3, 183–193 (2007). https://doi.org/10.1007/s11302-006-9027-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-006-9027-x