
Abstract
Gene flow in plant populations is heavily affected by species sexual systems. In order to study the effect of sexual systems on genetic structure, we examined plastid and nuclear DNA of 12 dioecious (males and females) and 18 trioecious (males, females and hermaphrodites) populations of Salix myrsinifolia—a boreal shrub with slow range expansion. Populations were located along latitudinal gradients across submarginal and marginal parts of the range. Individuals of each sex morph were all hexaploid. We identified 10 chloroplast DNA haplotypes and scored 205 polymorphic bands with amplified fragment length polymorphism. We found dioecious populations that differed from trioecious populations via the presence of four unique haplotypes and significant difference in Nei’s gene diversity index (0.119 vs. 0.116) and down-weighed marker value (1.17 vs. 1.02). The latter parameter, together with haplotype and nucleotide diversity, significantly decreased with latitude similar to the expansion front. Also, we found that 89% of hermaphrodite individuals belong to one distinct in tree parsimony network haplotype. This frequency significantly decreased with latitude towards the expansion front. We suspect that the presence of hermaphrodites in trioecious populations may represent a trade-off between the possibility of producing progeny by single hermaphrodites and genetic variability loss through autogamy. S. myrsinifolia benefits from trioecious sexual systems under colonization events. This phenomenon is no longer a gain closer to the core of the species range.
Similar content being viewed by others

Introduction
Patterns of species’ geographic range and genetic diversity are strictly shaped by life history traits such as reproduction type, breeding system(s) and dispersal mechanisms as well as natural processes or human impacts (e.g. isolation, fragmentation, climate changes) (Hamrick and Godt 1996; Barrett and Husband 1990; Gitzendanner and Soltis 2000; Nybom 2004). In order to disperse, species harness evolutionary potential and adopt various strategies enabling survival, reproduction and geographic expansion. Phenotypic variation positively affects adaptive potential of expanding species and the source of this plasticity lays in genetic variation. Different patterns of genetic variation during range expansion can be found both in theoretical assumptions and experimental studies. In a comprehensive review (134 studies and 115 species), Eckert et al. (2008) detected a decrease in genetic diversity in 64.2% of species. There was an increase in genetic differentiation in 70.2%. During human-mediated introductions, newly established populations most often suffered losses of allelic richness and heterozygosity, but rarely also underwent significant increase in both parameters (Dlugosch and Parker 2008). Recently, more attention has been paid to the consequences of natural, contemporary range expansions (Banks et al. 2010; Garroway et al. 2011; Swaegers et al. 2013). These studies showed that the current range expansion may not only generate more independently replicated edge populations but also likely results in different population genetic patterns throughout the expansion front. Those differences may derive from population-specific features.
In plants, one species may be characterized by different sexual systems. Sex determines the gene flow direction and rate. In this context, an interesting and poorly studied problem is the relationship between genetic diversity and phylogeographic patterns of plants characterized by different sexual systems—especially in the course of expansion to new territories. In plant species characterized by polymorphic sex, we expect spatial separation of two or three sex morphs. Apart from well-known dioecy, females and/or males may persist with hermaphrodites in either gynodioecious (pistillate and perfect flowers on different plants), androdioecious (staminate and perfect flowers on different plants) and/or trioecious (staminate, pistillate and perfect flowers) populations, respectively (Sakai and Weller 1999).
In dioecious populations, the two sexes are separated fully into male and female individuals, and cross-pollination is the only possible option (Vamosi et al. 2003). In other polymorphic sexual systems, both obligate outcrossing (females) and mixed mating (hermaphrodites) may occur. In plant species, sexual systems affect their ability to establish new populations. In the case of dioecy, females and males may be less likely to colonize unoccupied habitat than plants from hermaphroditic populations. This often requires only one individual to establish new populations (Pannell and Barrett 1998). This concept was already given by Baker (1955, 1967), who stated that under long-distance dispersal, self-compatible individuals are more likely to establish new populations than self-incompatible ones (so-called Baker’s law). This law can be extrapolated to the general context of colonization events (Pannell and Barrett 1998).
Importantly, sexual systems influence the genetic diversity at the population and species level (Dorken et al. 2002; Obbard et al. 2006; Korbecka et al. 2011). In entomogamous, hermaphroditic colonizers, the founding individuals are often isolated by space and pollinators (avoiding small and sparse plant populations). This could also favour uniparental reproduction (Barrett et al. 2008). Such possibility enables population development, but at the cost of inbreeding. Indeed, genetic variation is most often reduced, i.e. in introduced vs. native parts of the range (Kliber and Eckert 2005, Lin et al. 2009). Despite this disadvantage, most invasive species in South Africa were capable of uniparental reproduction including 100% of surveyed woody species (Rambuda and Johnson 2004). This finding is surprising, because woody species are largely long-lived and therefore have shown higher chance for outcrossing during their lifetime or can wait out unfavourable conditions. This assumption is supported by high ratio of dioecy in trees (i.e. van Dulmen 2001; Gross 2005) comparing to other angiosperms (Renner 2014). Thus, we suspect that departure from dioecy may be induced in woody species by other benefits during colonization phase. The most important benefit is availability of uniparental reproduction (selfing), but there is also a higher probability of cross-pollination and higher fruit sets.
Obbard et al. (2006) conducted a unique study on the relation between polygamic sexual system and genetic diversity patterns in the annual plant Mercurialis annua. This spans a range of systems from self-compatible monoecy through androdioecy to dioecy. The authors assumed that monoecy would be favoured during colonization, dioecy under stable conditions and androdioecy in intermediate ones. They showed that monoecious populations of this species displayed lower within-population genetic diversity than androdioecious populations and higher genetic differentiation than dioecious and androdioecious populations. This fits the assumption on the ‘colonization’ role in monomorphic individuals in plants capable of reproduction via different sexual systems. In turn, Dorken et al. (2002) showed a slightly lower (but not statistically significant) genetic variation between monoecious and dioecious Sagittaria latifolia populations. The substantial levels of selfing and inbreeding depression were expected in monoecious species but not dioecious ones. They proposed that sexual systems were maintained as the result of habitat differentiation and life history strategy.
The phylogeographic studies of plant species that exhibit variation in sexual systems are extremely scarce, but they explain the important way in which evolution of plant polymorphic sexual systems relates to patterns of colonization and extinction in the metapopulation sense (Dorken and Barrett 2004; Alonso and Herrera 2011). These authors stressed the origin of a given sexual reproductive system. For example, dioecy could be a result of historical factors and could be evolved from monoecy (Dorken and Barrett 2004; Panell and Dorken 2006). Similarly, hermaphroditism emerged as the most likely condition from gynodioecy during two events (Alonso and Herrera 2011). Furthermore, these studies, similar to Obbard et al. (2006), unravelled the metapopulation hypothesis for the distribution of sexual system.
Trioecy has received scant attention in population biology and genetic diversity studies because of its rarity in nature (3.6% of flowering plant species, Richards 1997; Albert et al. 2013) as well as difficulties with its identification and complexity. In the context of these unresolved issues, the large, cosmopolitan Salix genus provides an excellent biological system to study phylogeographic and population genetics. They enable a better understanding of the relationship between polymorphic sexual systems in the context of geographic expansion. The majority of species from this genus are dioecious, but at least 18 hermaphroditic, monoecious or trioecious species were noted (Mirski 2014).
One of the polygamic species with co-existing dioecy and trioecy populations is the hexaploid dark-leaved willow, Salix myrsinifolia (Faliński 1998, Mirski and Brzosko 2015, Mirski 2016). It is a boreal Eurasian, entomogamous and anemochorous shrub or small tree (Meikle 1984; Mirski and Brzosko 2015). This species was rare in Poland at the beginning of the twentieth century (Skvortsov 1968; Chmelař and Meusel 1986); but a few decades later, it spread in the south and west of Poland, often on anthropogenic habitats (Zieliński 1976, Faliński 1998, 2004, Sugier et al. 2011).
Faliński (1998, 2004) studied S. myrsinifolia populations in Lithuania and Poland and herbaria specimens across Europe and found that dioecious populations occurred in central and submarginal parts of the range. The trioecious populations were established across the border of Eastern Poland (Fig. 1a). He suspected that S. myrsinifolia hermaphrodites are characterized by bigger size, faster growth and higher reproductive potential. However, Mirski and Brzosko (2015) showed that hermaphrodites had intermediate values of morphological and reproductive traits between male and female individuals. They further showed that species with high reproductive potential (regardless of sex) produced hundreds of catkins containing several dozens or over hundreds of flowers. The sex structure of S. myrsinifolia populations showed a north/south gradient (Lithuania-Poland) at the periphery of distribution with female-biased structure in the north and balanced in the south. Furthermore, there was an increase in the frequency of hermaphrodites in recent decades, but only in the populations in the southwestern periphery of the species’ range (Mirski 2016).

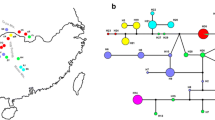
Geographic range of Salix myrsinifolia in Europe according to Chmelař and Meusel (1986) supplemented with local data from Zieliński (1976) and Sugier et al. (2011). a Distribution and frequency of plastid DNA haplotypes (unique haplotypes showed with red font, T trioecious pop., D dioecious pop.); b Bayesian consensus tree derived from 10 cpDNA sequences from trnL, trnT-trnL and accD-psaI intergenic spacers (bootstrap support >50% are given by the corresponding branches) (c) and statistical parsimony network (d)
Given the potential of S. myrsinifolia to expand its European geographic range (north/south direction), we studied the consequences of sexual systems on phylogeography and population genetic structure using chloroplast DNA (cpDNA) and nuclear molecular markers (amplified fragment length polymorphism, AFLP). We considered the polyploidy, dioecy/trioecy, dynamics of geographic range in last few decades and presence of hermaphrodites in S. myrsinifolia. We hypothesise that (i) genetic diversity of S. myrsinifolia is high because it is a polyploid with high sexual reproductive potential; (ii) dioecious populations maintain higher genetic diversity than trioecious ones in Poland because of the obligatory outcrossing; and (iii) the occurrence of hermaphrodites in S. myrsinifolia trioecious populations at the southern periphery of range is linked to a restricted subset of the cpDNA haplotypes vs. dioecious populations; and (iv) hermaphroditism occurring in the southern part of the European range is not an ancestral trait within this species but is an evolutionary transition from dioecy.
Methods
Sample collection
Thirty (12 dioecious and 18 trioecious) S. myrsinifolia populations were studied previously by Faliński (1998, 2004, see Introduction). These were selected along the north/south (Lithuania-Poland) latitude gradient (Fig. 1a, b; Table 1) at the periphery of geographic range. Leaf samples were collected from 4 to 20 individuals per population (depending on population size). The leaves were taken from individuals at least 2 m apart to avoid collecting clones. Where possible, samples were gathered randomly at corresponding to each population sex structure (ratio between males, females and hermaphrodite). If number of hermaphrodites was less very low (<3 individuals), all were taken. The 339 samples (132 from males, 146 from females and 61 from hermaphrodites) were studied with genetic analyses (Table 1).
Flow cytometry measurements
The genome size was estimated in fresh and young leaves of S. myrsinifolia that were collected from eight random trioecious populations. The flow cytometry samples were prepared according to Galbraith et al. (1983) with minor modifications. Plant tissues were diced with a sharp razor blade in a plastic Petri dish with 1 mL of Galbraith’s buffer [45 mM MgCl2, 30 mM sodium citrate, 20 mM 3-[N-morpholino] propanesulphonic acid, 0.1% (v/v) Triton X-100, pH 7.0] supplemented with propidium iodide (PI, 50 μg/mL) and ribonuclease A (50 μg/mL). The nuclei suspension was passed through a 50-μm mesh nylon filter. For each sample, 7000–10,000 nuclei were measured using Partec CyFlow SL Green flow cytometer (Partec GmbH, Münster, Germany) equipped with a high-grade solid-state laser with green light emission at 532 nm and side (SSC) and forward (FSC) scatters. Analyses were performed on 25 replicates for male, 37 for female and 28 for hermaphrodites of plant tissue over 3 days. Plants were raised in a growth chamber adjusted to a 16-/8-h, 26/20 °C day/night cycle. Leaves of Vicia villosa ‘Minikowska’ (2C = 3.32 pg; Dzialuk et al. 2007) were used as the internal standard. The histograms were analysed using the FloMax software (Partec, Münster, Germany). Means and standard deviations (SD) of the nuclear DNA content (in picograms, pg/2C) and nuclear DNA fluorescence index (DI = 2C of S. myrsinifolia/2C of V. villosa) of each of the three sex morphs of S. myrsinifolia were calculated. The genome size was estimated using the linear relationship between the ratio of target species and the internal standard 2C peak positions on the histograms. The mean coefficients of variation of the 2C DNA content were estimated for all S. myrsinifolia samples. The 1C genome size were obtained after converting values in picograms into base-pair numbers using the factor 1 pg = 978 Mbp (Doležel et al. 2003). Ploidy level was designated by comparing the samples to diploid standard of other species of the same genus - Salix caprea (DI = 0.220).
DNA extraction, cpDNA sequencing and data analyses
DNA was extracted from 0.2 g of leaf tissue with a Genomic Mini AXE Plant kit (A and A Biotechnology, Poland) according to the manufacturer’s instructions. Three fragments out of 15 non-coding and coding regions of chloroplast DNA (cpDNA) were analysed. Polymorphism was observed in the intron of the chloroplast trnL (UAA) gene and intergenic spacers of trnT-trnL (Taberlet et al. 1991) and of accD-psaI (Small et al. 1998). Fragments were amplified by PCR in a 10-μL volume containing 2 μL of DNA; 5 μL of QIAGEN Multiplex PCR Master Mix (with HotStarTaq® DNA polymerase, QIAGEN); 2 μL of H2O and 1 μL of 2 pmol of each primer. The PCR profiles comprised the following: initial denaturation at 94 °C for 15 min, 30 amplification cycles of denaturation at 94 °C for 30 s, annealing at 57 °C for 1 min and 30 s, extension at 72 °C for 1 min and a final extension of 30 min at 60 °C. Sequencing was performed using BigDye Terminator V 3.1 (Applied Biosystems) according to the manufacturer’s instructions. Samples were run on an ABI 3130 Genetic Analyser (Applied Biosystems). Sequences were assembled and edited using BIOEDIT 7.04 (Hall 1999).
Nucleotide (π) and haplotype (h) diversity, and Tajima’s D (Tajima 1989), Fs (Fu 1997) and R 2 (Ramos-Onsins and Rozas 2002) statistics were calculated with DNASP 5.0 (Librado and Rozas 2009). Ramos-Onsins and Rozas (2002) demonstrated that Fs and R 2 statistics have the greatest power to detect population expansion for non-recombining regions of the genome under a variety of circumstances especially when the population sample sizes are large (~50, Fu’s Fs) or when sample sizes are small (~10, R 2). They also found that the power of the R 2 statistic is relatively high when the number of segregating sites is low (e.g. ~20). The significance of Fu’s Fs and R 2 was obtained by examining the null distribution of 1000 coalescent simulations of these statistics using DNASP 5.0. The significantly negative Fu’s Fs and significantly positive R 2 values suggest population expansion. Quadratic regression in MS Excel (formula written using Visual Basic language) was performed to describe the relation between haplotype diversity and the frequency of a particular haplotype in the latitude gradient. To test the phylogeographic structuring of the haplotypes, the G ST (based on haplotype frequencies) and N ST (based on haplotype frequencies and genetic distance between haplotypes; Pons and Petit 1996) were calculated using DNASP 5.0. A chi-square test was applied to pairwise comparisons of haplotype frequency data among populations.
To investigate the relationships between haplotypes, phylogenetic analyses were carried out using a Bayesian approach implemented in BEAST 2.0 (Drummond et al. 2012) with Populus tremula as an outgroup. This showed that the Populus genus belongs to a clade having a sister relationship to the Salix genus (Cronk et al. 2015). We used the Beauti program to unlink the substitution models of the data partitions and to implement the models of sequence evolution identified as optimal by JMODELTEST 0.1.1 (Posada 2008). The model fit of nucleotide substitution models was assessed via the Akaike Information Criterion (AIC). Simulations were run using the Markov Chain Monte Carlo (MCMC) for 10,000,000 generations with a store of 10,000 in the BEAUTI program. Finally, we discarded the trees as burn-in and summarized the trees using TREEANNOTATOR 1.7.2; trees were visualized with FIGTREE 1.3.1 (Rambaut 2010). Bootstrap values >50% and posterior probabilities >0.95 were defined as evidence for supported relationships. We also constructed haplotype networks for each sequence using TCS 1.21 (Clement et al. 2000) to obtain a non-bifurcating perspective of relationships. We used the default settings with a 95% connection limit.
AFLP procedure and data treatment
The AFLP procedure followed Vos et al. (1995), but was modified according to the Applied Biosystems protocol (AFLP™ Plant Mapping). First, the 16 primer pair combinations were tested on four selected samples. The fluorescence-labelled selective amplification products were mixed with 500 Liz-labelled size standards (Applied Biosystems) and were run on an ABI 3130 Genetic Analyser. From this analysis, two primer combinations (EcoR1-AGC/MseI-CAT and EcoR1-AGG/MseI-CAC) that gave polymorphic, clear, reproducible fragments of homogeneous intensity were chosen. Variable fragments in the 70–500 bp size range were scored as present (1) or absent (0) using GENEMAPPER 4.0 (Applied Biosystems). To test the repeatability of AFLP results, two individuals from each population were completely replicated starting from the restriction/ligation reaction of AFLP. Potential resampling of clones was checked with AFLPDAT R-SCRIPT (Ehrich 2006).
We studied Nei’s gene diversity (Hj), proportion of polymorphic loci (PPL, AFLPSURV 1.0, Vekemans 2002), down-weighed marker value (DW, AFLPDAT R-SCRIPT, Ehrich 2006), and the Shannon diversity index (I, POPGENE 1.31, Yeh et al. 1999) in populations across the north/south gradient. These were tested for correlation with latitude.
The populations differed in the number of individuals and the ratio of sexes. Therefore, the number of samples could influence the genetic diversity indices (including sexual system comparison). To overcome this bias, we conducted resampling using R Software. We randomly drew six individuals from each population—two of each possible sex morph. We complemented the lacking samples with random-sex individuals in dioecious populations or trioecious where the number of particular sex morphs was below two. We obtained 180 elements in the dataset and used them to calculate AFLP genetic diversity parameters. After 1000 iterations, the mean parameter value and its standard deviation were calculated. The Kendal Tau coefficient was used to test if data pre- and post-resampling were correlated.
We assumed that the population size (N), latitude (Lat), hermaphrodite ratio (H R), forest cover (F COV) and open landscape cover (O COV) might impact the population genetic diversity. The F COV was assumed as a possible barrier to gene flow in this entomogamous species. It was measured in ArcGIS 10.0 software (upon Corine Land Cover 2006 data) as ratio of all forests over a 5.5-km radius around the populations. The distance used here was assumed to be a reasonable length to overcome a common pollinator like honey bees (Beekman and Ratkiens 2000; Hagler et al. 2011). The O COV represents possible habitats for S. myrsinifolia and better conditions for pollinator movement. This supports cross-pollination and gene flow. The Pearson correlation was used to create a matrix between genetic diversity indices and selected variables. The Spearman rank correlation was used only with DW because it differed from normal distribution (Statistica 10.0).
To infer population structure and assign individuals to populations, the model-based clustering method described by Pritchard et al. (2000) was used as implemented in STRUCTURE 2.3.4. The AFLP datasets were coded with a top row indicating 1 as the recessive allele in STRUCTURE 2.3.4 including studies using dominant markers (Falush et al. 2007). Data were analysed with an admixture model with correlated allele frequencies elaborated by Falush et al. (2003). Ten replicates were run for all possible values of the number of clusters (K) up to K = 15 (Online Resource 2). Following the recommendations by Evanno et al. (2005), the ad hoc statistic ΔK, based on the rate of change in the log likelihood of data between consecutive K values, was calculated. All runs were based on 500,000 iterations after a burn-in of 100,000 iterations.
We investigated the population genetic structure among sample locations calculating pairwise F ST values and testing their significance with Fisher’s exact tests after 10,000 permutations as implemented in ARLEQUIN 3.11 (Excoffier et al. 2005). The partitioning of variation at different levels (the whole data set and dioecy and trioecy population groups, respectively) was calculated by analysis of molecular variance (AMOVA) in ARLEQUIN 3.11 using 1000 permutations. The relationships between genetic and geographic distance (isolation by distance, IBD) among all studied populations were estimated by correlating F ST/(1 − F ST) with ln geographic distance (km) in a Mantel test using GENALEX 6 (Peakall and Smouse 2006); 9999 random permutations were studied. Genetic relationships between individuals were identified by principal coordinate analysis (PCoA, based on Jaccard similarities) computed in PAST 2.01 (Hammer et al. 2001) and visualized in Statistica 10.0 (StatSoft Inc.). The differences in average values of PC1 and PC2 between individuals were tested by one-way Anova.
Past, present and future species distribution models
Range expansion of S. myrsinifolia on south-western edge of its distribution is poorly documented; therefore, we carried out predictive species distribution models (SDM) in order to confirm and follow species expansion. SDMs were done for the past, present and future under bioclimatic variables. Modelling was done in MaxEnt 3.3.3k software (Phillips et al. 2006). Species occurrence data was computed from a few sources: the Global Biodiversity Information Facility (GBIF) database (2016), published data on species distribution range and 30 self-studied populations. The GBIF database covered mostly northern and western Europe and therefore, the dataset had to be reduced and completed with other locations in eastern Europe. Therefore, 500 localities were randomly drawn from the GBIF database, and 500 points were also randomly marked (in Quantum GIS 2.10 software) inside European species distribution according to Chmelař and Meusel (1986). The final dataset covered all European species range with nearly equal density. Thus, the model was not influenced by an unproportional number of data from different areas.
Climatic variables derived from the CMIP5/PMIP3 multi-model ensemble were downloaded from ecoClimate database (Lima-Ribeiro et al. 2015) over four periods: pre-industrial (piControl-1760), historical (1900–1949), modern (1950–1999) and future (2080–2100). The climate change scenario used for the projections included two representative concentration pathways (RCPs) reported in the IPPC5 data: RCP 8.5 (~8.5 W m2 stabilized after 2100, slightly more severe than SRES A2) and a stronger mitigation scenario RCP 4.5 (~4.5 W m−2 stabilized after 2100, which is similar to the lowest-emission scenarios (B1) assuming the imposition of a series of emission-mitigation policies. In all periods/scenarios, we used all of the accessible ecoClimate database models and averaged the output habitat suitability raster in ArcGIS 10.0. Future data included the following global climate models (GCM): CCSM, CNRM, FGOALS, GISS, IPSL and MIROC. The past and present data also included MPI and MRI models.
First, the present climatic data (19 variables) were extracted for species occurrence points in ArcGIS 10.0 and were inter-correlated. From the Pearson correlation matrix, we picked all the variables that were not correlated with any other with correlation coefficient greater the 0.7 including annual mean temperature (BIO1), mean diurnal temperature range (BIO2), isothermality (BIO3), max temperature of the warmest month (BIO5), temperature annual range (BIO7), annual precipitation (BIO12) and precipitation seasonality (BIO15).
The habitat suitability model for ‘modern’ conditions was built in MaxEnt and has been projected to other periods. To validate the model, occurrence points were divided into two sets that train the model (75%) and test it (25%). The logistic output in MaxEnt estimates the probability of presence assuming that the sampling design is such that typical presence localities have a presence probability of ca. 0.5; however, this value is fairly arbitrary (Liu et al. 2005; Phillips et al. 2006). Therefore, we selected those catchments corresponding to MaxEnt values indicating maximum training sensitivity plus specificity. This is a more robust approach (Liu et al. 2005).
Results
Nuclear DNA content and ploidy level
The genome size estimated for males, females and hermaphrodites of S. myrsinifolia was 2.172, 2.164 and 2.160 pg/2C DNA, respectively, with no significant differences between these groups (Online Resource 1). The average nuclear DNA content for this species was 2.165 pg/2C, which corresponds to 1059 Mpb. The resulting histograms were good quality with mean CVs between 3.83–4.07% for the target (Supplementary information Table S1, Fig. S1). The internal standard, V. villosa, presented a mean coefficient of variation (CV) value of 3.40%. The mean nuclear DNA fluorescence index of all studied samples ranged from 0.651 to 0.654 (Online Resource 1). Nuclear DNA shows that all examined individuals (regardless of sex and population) were hexaploid.
Plastid DNA analysis
The lengths of the trnL and trnT-trnL and of accD-psaI were 449, 354 and 588 bp, respectively. The concatenated alignment was 1264-bp long. The 315 individuals analysed for three regions yielded 10 cpDNA haplotypes. Of these, six (H1–H6) were noted for dioecious and trioecious populations that were shared by at least two populations. Four were unique (H7–H10) and appeared only in dioecious populations (Fig. 1a, b) and only in the submarginal part (Lithuania). The dominant haplotypes (H1 and H2) were found in 80 and 90% of all populations, respectively, while other common types were shared among 13–56% of all populations.
Each population consisted on average of 3.2 haplotypes. Dioecious populations contained insignificantly more haplotypes than trioecious populations (3.6 vs. 2.9, U = 137, p = 0.113). Haplotype diversity (h) reached on average 0.57 (0.60 and 0.55 in dioecious and trioecious populations, respectively). Nucleotide diversity (π) reached 0.90 × 10−3; 0.98 in dioecious and 0.85 in trioecious populations (Table 1). Neither parameter differed between groups (U = 117, p = 0.719 and U = 123, p = 0.542, respectively). There was a marginally significant spatial pattern for h in the latitude gradient (r 2 = 0.13, p = 0.05, Fig. 2a). Estimates of Tajima’s D (0.358), Fu’s Fs (−0.654) as well as R 2 statistic (0.078) were not significant (p > 0.05). The observed mismatch distribution also differed from sudden range expansion (Online Resource 2). The total N ST (0.205) was nearly the same as G ST (0.206) indicating a weak phylogeographic structuring of the haplotypes.

Bayesian clustering results for Salix myrsinifolia populations (for K = 2, each individual is represented as vertical bar partitioned into K coloured segments) generated by STRUCTURE software from AFLP markers data. Here, T and D stand for trioecious and dioecious populations, respectively
The most frequent haplotypes (H1 and H2) were distributed both in dioecious and trioecious populations (Fig. 1b). Haplotype H1 was noted in 45% of females, 43% of hermaphrodites and 12% of males (Fig. 3). Other common haplotypes (H2, H3) were more balanced in males and females; the hermaphrodites were accessory (<3%) (Fig. 3). The haplotype H1 was found in 89% of hermaphrodites and distributed among every trioecious S. myrsinifolia population. In addition, the haplotypes H2, H3 and H5 were shared between the remaining hermaphrodite individuals (1, 2 and 8%, respectively). Only the frequency of haplotype H1 had a significant increase in the south (r 2 = −0.39, p < 0.001, Fig. 2b).

Frequency of sex morphs among most common Salix myrsinifolia haplotypes (H1, H2 and H3)
The strict consensus tree revealed one well-supported S. myrsinifolia haplogroup with 100% posterior probabilities (Fig. 1c). Within this group, unique haplotypes H9 and H10 as well as H8 and H7 formed two separate and well-supported subgroups (62 and 100% bootstrap support, respectively); haplotypes H3 and H6 occurred in the third subgroup (99% bootstrap support) (Fig. 1c). The network tree resulted in a star-like shape with the most frequent haplotypes H2 in the centre of the network tree surrounded by remain haplotypes with lower frequency. The haplotype H1 represented almost all hermaphrodites in the southern part of the species’ range with a similar high frequency to H2. This was observed on the edge of the parsimony network (Fig. 1d).
Nuclear DNA analysis
Two hundred and five polymorphic bands were scored with AFLP. The average genotyping error rate was estimated at 3.4%. The results prior to and after resampling were significantly and positively correlated. Coefficients of Kendall’s tau reached 0.406 for PPL, 0.875 for Hj, 0.783 for I and 0.862 for DW. Differences prior to and after resampling were statistically significant in all studied parameters: PPL (H = 9.1, p = 0.0025); Hj (H = 4.3, p = 0.038); I (H = 11.4, p = 0.0007) and DW (H = 34.8, p < 0.0001). Values of above genetic indices after resampling were relatively moderate and reached slightly higher values in the dioecious than in the trioecious populations (Table 1). The mean percent of polymorphic loci was 32.8 ± 3.2%, and there was no significant difference in PPL values between the dioecious (33.4%) and trioecious populations (32.4%, Mann–Whitney test, U = 126.0, p = 0.281). We found significant differences in the total Nei’s gene diversity (Hj) between dioecious and trioecious populations (0.119 vs. 0.116; U = 620, p = 0.039), but not in the Shannon index (I) (0.121 vs. 0.118; U = 87, p = 0.384). The mean values of those indices were 0.117 and 0.119, respectively. The frequency down-weighed marker value (DW) equalled 1.08 ± 0.27 and differed significantly between dioecious and trioecious populations (1.17 vs. 1.02; Mann–Whitney test, U = 151, p = 0.036).
The correlation matrix between genetic diversity indices and population size, latitude, ratio of hermaphrodites and afforested and open habitats showed few significant correlations (Online Resource 3). The population size was significantly and negatively correlated with h (r = −0.50, p = 0.02) and π (r = −0.36, p = 0.024), and latitude was significantly and negatively correlated with DW (r = −0.46, p = 0.048), but positively with π (r = 0.32, p = 0.044).
Stepwise clustering performed in Structure separated individuals into two groups (Online Resource 4). At K = 2, clusters were not distinguished geographically or genetically from each other, but specimens from almost all populations represented a mixture of two diverse genetic backgrounds. Two gene pools were balanced in all populations but these were slightly dominated by one gene pool (green) at the southern periphery of the range (Fig. 4).

Quadratic regression between Salix myrsinifolia haplotype diversity (a) and frequency of H1 haplotype (b) and latitude (b)
The populations were weakly differentiated (F ST = 0.064, p < 0.001) in this part of its European range. The Mantel test revealed a significant correlation between genetic and geographic distances (Fig. 5, r 2 = 0.024, p = 0.025). Analysis of molecular variance (AMOVA) based on all populations indicated that the majority of genetic variation (93.7%) occurred within populations, while the variation between populations was 6.3%. AMOVA-based on dioecy and trioecy group division gave a similar result (93.6% variation within populations, 6.2% variation among populations within sexual systems, 0.2% among sexual systems groups) (Table 2).

Isolation by distance pattern of Salix myrsinifolia in north/south gradient of the studied part of the geographic range (Lithuania-Poland)
The PCA indicated that male, female and hermaphrodite individuals from populations of different sexual systems were mixed together on the PCoA diagram plot. The first two components explained 15.4 and 8.8% of the variance. Females, males and hermaphrodites were not grouped together in relation to sex (Fig. 6).

Principal component analysis (PCoA) showing Jaccard’s genetic similarity among 339 individuals of Salix myrsinifolia (squares males, circles females, triangles hermaphrodites; white marks dioecious and black trioecious populations)
Species distribution model
The MaxEnt model of climatically suitable areas showed high reliability when training data were compared to a test subset of occurrence points—the AUC value reached 0.966. The jacknife test revealed that the annual mean temperature (BIO1), mean diurnal temperature range (BIO2) and maximum temperature of the warmest month (BIO5) had the greatest impact on the model training. Models illustrated a climatically suitable area that slowly spread southwards in the last centuries (ca. 80 km/century) (Fig. 7). However, future projection of the model shows a rapid decline of habitat suitability—not only in this area, but also in most parts of Central Europe, both under RCP 4.5 and 8.5 scenarios (Online Resource 5). At the end of the twenty-first century (2080–2100), Fenno–Scandia, Northern Russia and parts of the Alps and Scotland may remain climatically suitable.

Borders of the optimal climatic suitability areas (suitability index >0.5) for Salix myrsinifolia in different periods modelled with MaxEnt software. Models built under seven bioclimatic variables (BIO1, BIO2, BIO3, BIO5, BIO7, and BIO12) from eight different global climate models (CCSM, CNRM, FGOALS, GISS, IPSL, MIROC, MPI, MRI) and averaged
Discussion
Hermaphroditism is a rather rare phenomenon in regularly dioecious Salicaceae family, and S. myrsinifolia is an interesting model species to investigate the relationship between sex and genetic structure. For the first time, we report here the genome size of this species (2.165 pg/2C) as well as for any hexaploid species in the Salix genus (Online Resource 1). We found hexaploids in each of the individuals investigated with flow cytometry, confirming the results of Håkansson (1955), Dreschler (in Neumann 1981) and Buechler (2010) concerning the ploidy level in this species and supporting our assumptions connected with the ecological and molecular consequences of the polyploidy level and the methods used to study it (Comai 2005, Birchler 2012).
Genetic variation in willows is quite high vs. other woody species (Lin et al. 2009). This can be attributed to obligate outcrossing in dioecious populations, anemochory (contributing to long-distance dispersal) and the general commonness of individuals (supporting the probability of cross-pollination). We found that S. myrsinifolia has relatively moderate genetic diversity (PPL = 32.8; Hj = 0.117, I = 0.119), mostly lower than other Salix species, i.e. Salix herbacea (PPL = 96.5, Hj = 0.03–0.16; Alsos et al. 2009); Salix daphnoides (diploid, PPL = 55.5; Sochor et al. 2013) or Salix phylicifolia (PPL = 56–75; Egelund et al. 2012). Both haplotype and nucleotide diversity (h = 0.57; π = 0.90 × 10−3) were relatively lower than in Salix arctica (tetraploid, h = 0.77–0.80); Salix viminalis (diploid, π = 6.55 × 10−3) and Salix schwerinii (diploid, π = 8.49 × 10−3) (Steltzer et al. 2008, Berlin et al. 2011), but there are only a few studies of plastid DNA inside this genus for comparison. In other members of Salicaceae, genetic diversity was usually also higher, i.e. Populus nigra (Hj = 0.139–0.183; Smulders et al. 2008) and Populus alba (PPL = 39–72, I = 0.239–0.327; Castiglione et al. 2010). One surprising finding is that h and π were negatively correlated with population size. This is opposite of the widely acknowledged principle of positive relation between population size and genetic variation (Frankham 1996). In our case, a few populations proved to be the most variable, although small in size (population nos. 6D, 7D, 8D, 21T, 22T; Table 1). Correlations between population size, h and π were no longer significant when those populations were excluded. As mentioned, small populations are located in two groups (Fig. 1b). This is possibly supported by longitudinal expansions; therefore, the reverse relationship between plastid DNA variability and population size is most probably an artefact.
Overall, our hypothesis (i) on high genetic diversity of S. myrsinifolia was not supported by data gathered. Our assumption of high genetic diversity of this species was based on its polyploidy. Both theoretical and empirical studies showed that the ploidy level benefits genetic variance (Stebbins 1984, Luttikhuizen et al. 2007). But if polyploidy is a rather recent phenomenon, then it requires some time to ‘recover’ equilibrium between mutation and drift (Luttikhuizen et al. 2007). Such situations are likely under glaciation episodes that are would affect our species of interest. Relatively moderate genetic variance of S. myrsinifolia may result from bottlenecks during range contractions in glaciation episodes. Finally, we have studied only the marginal and submarginal parts of the range. In general, populations on the border of the range often suffer from lower genetic variation and episodes of range expansion and contraction. This might underlie the genetic variation (Lesica and Allendorf 1995, Eckert et al. 2008).
Genetic diversity patterns between dioecious and trioecious populations partially confirmed hypothesis (ii). Haplotype and nucleotide diversity were slightly higher in the first group. However, cpDNA variation revealed no significant differences between dioecious and trioecious populations. Moreover, higher AFLP diversity for dioecious than trioecious populations were observed, although only differences in Hj and DW were statistically significant (Table 1). However, the AMOVA results showed that the difference in variance between groups of different sexual systems was not strongly pronounced.
Similar comparison between marginal and submarginal parts of S. myrsinifolia range showed a spatial trend in haplotype diversity. The northern (submarginal; Lithuania) populations contained a higher number of haplotypes than the southern (marginal; East Poland) ones (Fig. 1b). These also included the only four unique haplotypes that are distinctive in the tree parsimony network (Fig. 1d). Moreover, there was a significant and decreasing spatial trend of both h (Fig. 2a) and π (Online Resource 3) in the south direction. The haplotype distribution along the north/south gradient showed the unimodal pattern in S. myrsinifolia. The peak level of the haplotype diversity occurred between submarginal and marginal populations. This interesting finding indicates that a bottleneck may occur under more recent range expansion of the S. myrsinifolia European range. This was confirmed by simulations of the climatically suitable area, which increased from pre-industrial–1760 to 1950–1999 periods (Fig. 7). Thus, low nucleotide diversity should be expected in a recent range expansion. Of the calculated nuclear DNA indices, the same spatial trend was significant only for DW (Online Resource 3). This showed a gradual decrease towards the south.
Literature survey and climatic models showed that S. myrsinifolia is currently expanding its range by about 0.8 km per year. This fits the velocity of 0.01–1.5 km/year expected for natural expansion of other plant species (Corlett and Westcott 2013). At the same time, studies on S. myrsinifolia (Mirski and Brzosko 2015, Mirski 2016) showed that trioecy in S. myrsinifolia is common on the fringes. It is advantageous at this location.
However, in this paper, we suggested that it might be disadvantageous (comparing with dioecy) in terms of genetic diversity (closely related to phenotypic variation). Therefore, we suspect that the distribution of dioecious and trioecious S. myrsinifolia populations may result from a trade-off between sexual reproduction and benefits from uniparental reproduction during range expansion. Several facts below indirectly confirm this hypothesis.
First, the distribution of hermaphrodites in S. myrsinifolia follows the expansion front (Mirski 2016), but there was no advantage of this sex morph over males and females other than better colonization properties of self-compatible hermaphrodites (Mirski and Brzosko 2015). Even if only one hermaphrodite individual appears at the new site, it is capable of seed set through selfing (Mirski, pers. observ.). On the other hand, selfing is known to negatively affect population genetic variation and hermaphroditism may no longer be an advantage when populations are well established. This is visible in the central parts of the species range, where Faliński (1998) examined S. myrsinifolia herbaria specimens and found hermaphroditism only as an exceptional phenomenon. Also, recent sex structure monitoring showed that the frequency of hermaphrodites decreased in the submarginal range, but increased in the marginal part; the male-to-female ratio was constant over time (Mirski 2016). Most probably, the frequency of hermaphrodites still increased closer to the moving expansion front, but decreased with the distance to its edge and time.
The adjustment of sex to environmental conditions (incl. spatial sex structure) is a common issue in plants, but is rare in dioecious species. Understandably, hermaphrodites are the most convenient to adjust; but in many dioecious plants, the mechanisms of sex determination are already quite consolidated. That is, the evolution of sex chromosome is an ongoing process in some plants (Vyskot and Hobza 2004), and thus the turnover is rare. However, this is the case of a few other taxa like Acer negundo (Mędrzycki et al. 2006), M. annua (Pannell et al. 2008), S. latifolia (Dorken and Barrett 2004) and Wurmbea dioica (Barrett et al. 1999). Also, in the Salix genus, in which sex is determined by one locus or a few (Pucholt et al. 2015), many species show sex-plasticity, but frequency of hermaphroditism is still very rare (Mirski 2014). Since all other sexual systems evolved from hermaphrodite ancestors (Charlesworth 2002), the drawback is still possible, even in species with already established chromosomes. A body of literature documents such sex changes in dioecious species and points out responsible environmental factors (Freeman et al. 1980).
The geographic patterns of willow genetic variation have not thus far been extensively described, but in general populations at the edge of distribution often shows lower values of variation than more central ones (Eckert et al. 2008). For example, in Salix arbutifolia, genetic variation was lowest in the most peripheral populations (He = 0.217 and 0.362 vs. 0.602 on average, Nagamitsu et al. 2014). In other species—S. daphnoides, also studied at the edge of its geographical distribution, genetic variation was moderate, but considerably higher in primary than in the secondary habitat (He = 0.323–0.618 vs. 0.125–0.375, Sochor et al. 2013). In contrast, the widespread S. viminalis showed higher genetic variability across Europe (mean He = 0.62), but also significant differentiation between studied regions. Also, populations from Western Russia, which are closer to the centre of the whole Eurasian range, were slightly more variable than those from more westwards parts of Europe (Berlin et al. 2014). High genetic variability was also shown in an Irish study of other widespread and common species—Salix caprea (mean He = 0.58), but rarer and spatially restricted subalpine species in Britain, like Salix lanata, Salix lapponum and S. herbacea also possessed high genetic variation (He = 0.706, 0.703 and 0.527, respectively) (Stamati et al. 2007). All of these examples concern European species of similar reproductive strategies (dioecious, entomogamous and anemochorous). The differences in their genetic variability values show that their position in the geographic range influences genetic variability as well as other population-specific factors.
In dioecious species, genetic variation is heavily affected by the probability of gene exchange through seeds or pollen. Van Puyvelde and Triest (2007) showed low gene flow in S. alba that was restricted to distances of 1–3 km. On the contrary, Perdereau et al. (2014) reported a high gene flow in S. caprea to distances over 200 km. Thus, we cannot expect one pattern of genetic variation in large and variable Salix genus, which is capable of both sexual and asexual reproduction. In S. myrsinifolia, we found a gradual loss of h, π and DW (Fig. 2a, Online Resource 2) following the expansion front. We also found weak genetic structure but significantly isolated by distance. The populations were slightly differentiated, while genetic and geographic distances were significantly correlated. The plastid and nuclear DNA results illustrated a pattern of recent but not sudden stepping-stone expansion across investigated area.
Recent studies on sex determination in S. viminalis and S. suchowensis showed that sex chromosomes are in the early stage of divergence, and sex is determined by the ZW system, with female being the heterogametic sex (Hou et al. 2015, Pucholt et al. 2015). However, the sex determination system may still be unsteady in this species because hermaphroditism occurs in different Salix species (Mirski 2014) and S. myrsinifolia individuals are even capable of changing sex during their life cycle (Mirski 2016). This may be caused by a high turnover rate in sex determination mechanisms in the entire Salicaceae family (Pucholt et al. 2015). Hermaphroditism was so never previously mentioned in studies of Salicacae sex determination because it is rare. Our results are the first to deal with this issue. We show a distinct link between S. myrsinifolia genetic structure and hermaphroditism. Considering that ploidy levels did not differ between hermaphrodites and unisexual individuals (Online Resource 1), it is rather unlikely that hermaphroditism in this species could be derived from hybridisation with other willows species. There is, however, the chance of hybridization with other hexaploid willow, but according to our knowledge, there were no willows of the same ploidy level in the studied area. However, hybridization with hexaploid species or hybrids of unknown origin cannot be ruled out. We excluded different sexual systems in populations that resulted from different ploidy levels such as M. annua (Pannel et al. 2008). However, the spatial distribution of hermaphroditism (Mirski 2016) suggests a possible genetic mechanism for this phenomenon. This study revealed that hermaphroditism follows a spatial genetic structure that we assumed in hypothesis (iii). Only a few hermaphroditic individuals spread to other haplotypes (H2, H3 and H5), and 89% of hermaphrodites belonged to one haplotype (H1) (Fig. 3). Moreover, out of two dominant haplotypes (H1 and H2), only the ‘hermaphroditic’ one (H1) significantly increased the frequency towards the southern range margin in accordance with expansion front (Fig. 2b). Also, the tree parsimony network showed that the H1 haplotype is distinctive from the others.
In sum, this proves that hermaphroditism is bonded with a particular haplotype and follows its spatial distribution. The possible mechanism underlying the spread of hermaphroditism might be gene surfing, described in this context by Excoffier et al. (2009). The alleles responsible for hermaphroditism could originate anywhere and stayed at low frequencies as a neutral mutation. However, at the distribution edge, they became advantageous and were propagated by the expansion wave. We suspect that the main advantage of hermaphroditism is its ability to be the ‘best colonizer,’ because each individual is capable of producing seeds. The other haplotypes appear later, and the sexual structure is balanced at dioecy stage. In addition, S. myrsinifolia on the SW edge of its distribution most often inhabits the anthropogenic habitats. These are often under succession or anthropogenic changes (Faliński 1998, Mirski 2016). Therefore, its populations are rather short-lived (Mirski 2016). These features of a good colonizer are needed to last in unstable conditions.
Another question is the genesis of hermaphroditism in regularly dioecious species like willows. We hypothesised this trait is not-ancestral and could be expected from general patterns of sexual systems evolution (Barret et al. 2002) or even from particular cases of similar sex systems transitions, i.e. in the case of subdioecious Daphne laurelola (Alonso and Herrera 2011). Rather, we suspect that the hermaphroditism in S. myrsinifolia individuals is an evolutionary transition from dioecy to trioecy. The following facts support this hypothesis: haplotypes in trioecious populations are the restricted subset of those found in dioecious ones, and the genetic diversity is lower in trioecious than dioecious populations. This proves that dioecy is the ancestral condition to hermaphroditism in these species.
We showed that hermaphroditism is bonded with margins of S. myrsinifolia range and governed mostly by particular haplotype. An interesting question is: will this feature last under changes of distribution range and if so, where? SDM confirmed that climatic conditions have thus far supported range expansion southwards and westwards, but future projections show climatic conditions will get steeply worse for this species (Online Resource 5). Hermaphroditism should be still favoured as advantageous in low population density and under unstable conditions during the predicted range contraction. However, if conditions rapidly worsen, then the southern hermaphroditic ‘colonizer’ haplotype (H1) may be lost due to genetic drift.
References
Albert B, Morand-Prieur ME, Brachet S, Gouyon PH, Frascaria-Lacoste N, Raquin C (2013) Sex expression and reproductive biology in a tree species, Fraxinus excelsior. C R Biol 336:479–485
Alonso C, Herrera CM (2011) Back-and-forth hermaphroditism: phylogenetic context of reproductive system evolution in subdioecious Daphne laureola. Evolution 65:1680–1692
Alsos IG, Alm T, Normand S, Brochmann C (2009) Past and future range shifts and loss of diversity in dwarf willow (Salix herbacea L.) inferred from genetics, fossils and modelling. Glob Ecol Biogeogr 18:223–239
Baker HG (1955) Self-compatibility and establishment after "long-distance" dispersal. Evolution 9:347–349
Baker HG (1967) Support for Baker’s law as a rule. Evolution 21:853–856
Banks SC, Ling SD, Johnson CR, Piggott MP, Williamson JE, Beheregaray LB (2010) Genetic structure of a recent climate change-driven range extension. Mol Ecol 19:2011–2024
Barrett SCH, Husband BC (1990) The genetics of plant migration and colonization. In: Brown AHD et al (eds) Plant population genetics, breeding, and genetic resources. Sunderland, Sinauer & Associates, pp 254–277
Barrett SCH, Case AL, Peters GB (1999) Gender modification and resource allocation in subdioecious Wurmbea dioica (Colchicaceae). J Ecol 87:123–127
Barrett SCH, Colautti RI, Eckert CG (2008) Plant reproductive systems and evolution during biological invasion. Mol Ecol 17(1):373–383
Beekman M, Ratkiens FLW (2000) Long-range foraging by the honey-bee, Apis mellifera L. Funct Ecol 14:490–496
Berlin S, Fogelqvist J, Lascoux M, Lagercrantz U, Rönnberg-Wästljung AC, Karp A, Hanley SA (2011) Polymorphism and divergence in two willow species, Salix viminalis L. and Salix schwerinii E. Wolf G3(1):387–400
Berlin S, Trybush J, Fogelqvist J, Gyllenstrand N, Hallingbäck HR, Åhman I, Nordh NE, Shield I, Powers SJ, Weih M, Lagercrantz U, Rönnberg-Wästljung A-C, Karp A, Hanley SJ (2014) Genetic diversity, population structure and phenotypic variation in European Salix viminalis L. (Salicaceae). Tree Genet Genomes 10:1595–1610
Birchler JA (2012) Genetic consequences of polyploidy in plants. In: Soltis PS, Soltis DE (eds) Polyploidy and genome evolution. Springer-Verlag, Berlin Heidelberg, pp 21–32
Buechler WK (2010) Estimating polyploidy levels in fossil Salix: a critical review of cell size proxy methods. PaleoBios 29:60–75
Castiglione S, Cicatelli A, Lupi R, Patrignani G, Fossati T, Brundu G, Sabatti M, van Loo M, Lexer C (2010) Genetic structure and introgression in riparian populations of Populus alba L. Plant Biosyst 144:656–668
Charlseworth D (2002) Plant sex determination and sex chromosomes. Heredity 88:94–101
Chmelař J, Meusel W (1986) Die Weiden Europas: Die Gattung Salix. Die Neue Brehm-Bücherei 494. A. Ziemsen Verlag, Wittenberg Lutherstadt
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:657–1659
Comai L (2005) The advantages and disadvantages of being polyploid. Nat Rev Gen 6:836–846
Corlett RT, Westcott DA (2013) Will plant movements keep up with climate change? TREE 28:482–488
Cronk QCB, Needham I, Rudall PJ (2015) Evolution of catkins: inflorescence morphology of selected Salicaceae in an evolutionary and developmental context. Front Plant Sci 6:1030. doi:10.3389/fpls.2015.01030
Dlugosch KM, Parker M (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17:431–449
Doležel J, Bartos J, Voglmayr H, Greilhuber J (2003) Nuclear DNA content and genome size of trout and human. Cytometry 51A:127–128
Dorken ME, Barrett SCH (2004) Phenotypic plasticity of vegetative and reproductive traits in monoecious and dioecious populations of Sagittaria latifolia (Alismataceae): a clonal aquatic plant. J Ecol 92:32–44
Dorken ME, Friedman J, Barrett SCH (2002) The evolution and maintenance of monoecy and dioecy in Sagittaria latifolia (Alismataceae). Evolution 56:31–41
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evo 29:1969–1973
Dzialuk A, Chybicki I, Welc M, Śliwińska E, Burczyk J (2007) Presence triploids among oak species. Ann Bot 99:959–964
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical range: the central marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Egelund B, Pertoldi C, Barfod AS (2012) Isolation and reduced gene flow among Faroese populations of tea-leaved willow (Salix phylicifolia, Salicaceae). New J Bot 2:9–15
Ehrich D (2006) AFLPDAT: a collection of R functions for convenient handling of AFLP data. Mol Ecol Notes 6:603–604
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501
Faliński JB (1998) Androgyny of individuals and polygamy in populations of Salix myrsinifolia Salisb. In the south-western part of its geographical range (NE-Poland). Perspect Pl Ecol Evol Syst 1:238–266
Faliński JB (2004) Inwazje w świecie roślin: mechanizmy, zagrożenia, projekt badań. (Invasions in world of plants: mechanisms, threats, study draft). Phytocenosis 16:14
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7:574–578
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Freeman DC, Harper KT, Charnov EL (1980) Sex change in plants: old and new observations and new hypotheses. Oecologia 47:222–232
Fu XY (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E (1983) Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 220:1049–1051
Garroway CJ, Bowman J (2011) The genetic signature of rapid range expansion by flying squirrels in response to contemporary climate warming. Glob Change Biol 17:1760–1769
Gitzendanner MA, Soltis PS (2000) Patterns of genetic variation in rare and widespread plant congeners. Am J Bot 87:783–792
Gross CL (2005) A comparison of the sexual systems in the trees from the Australian tropics with other tropical biomes—more monoecy but why? Am J Bot 92:907–919
Hagler JR, Mueller S, Teuber LR, Machtley SA, Van Deynze A (2011) Foraging range of honey bees, Apis mellifera, in alfalfa seed production fields. J Insect Sci 11:144 available online: insectscience.org/11.144
Håkansson A (1955) Chromosome numbers and meiosis in certain Salices. Hereditas 41:454–482
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis. Available at: http://www.mbio.ncsu.edu/BioEdit/bioedit.html (accessed 10 October 2015)
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: Paleontological Statistics Software Package for education and data analysis Palaeontol Electron:4
Hamrick JL, Godt MJW (1996) Effects of life history traits on genetic diversity in plant species. Phil Trans R Soc B 351:1291–1298
Hou J, Ye N, Zhang D, Chen Y, Fang L, Dai X, Yin T (2015) Different autosomes evolved into sex chromosomes in the sister genera of Salix and Populus. Sci Rep 5:9076. doi:10.1038/srep09076
Kliber A, Eckert CG (2005) Interaction between founder effect and selection during biological invasion in an aquatic plant. Evolution 59:1900–1913
Korbecka G, Hamilton A, Pannell JR (2011) Mixed mating in androdioecious Mercurialis annua inferred using progeny arrays and diploid-acting microsatellite loci in a hexaploid background. Ann Bot 107:1057–1061
Lesica P, Allendorf FW (1995) When are peripheral populations valuable for conservation? Conserv Biol 9:753–760
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Lima-Ribeiro MS, Varela S, González-Hernández J, de Oliveira G, Diniz-Filho JAF, Terribile LC (2015) EcoClimate: a database of climate data from multiple models for past, present, and future for macroecologists and biogeographers. Biodivers Inform 10:1–21
Lin J, Gibbs JP, Smart LB (2009) Population genetic structure of native versus naturalized sympatric shrub willows (Salix; Salicaceae). Am J Bot 96:771–785
Liu C, Berry PM, Dawson TP, Pearson RG (2005) Selecting thresholds of occurrence in the prediction of species distributions. Ecography 28:385–393
Luttikhuizen PC, Stift M, Kuperus P, van Tienderen P (2007) Genetic diversity in diploid vs. tetraploid Rorippa amphibian (Brassicaceae). Mol Ecol 16:3544–3553
Mędrzycki P, Kołaszewska B, Browiński P (2006) Subdioecy in invasive populations of Acer negundo (Aceraceae) in eastern Poland. Polish Botanical Studies 22:355–364
Meikle RD (1984) Willows and poplars of great Britain and Ireland. Botanical Soc British Isles, London
Mirski P (2014) Exceptions from dioecy and sex lability in genus Salix. Dendrobiol 71:167–171
Mirski P (2016) Spatio-temporal dynamics of sex structure in trioecious and dioecious populations of Salix myrsinifolia. Folia Geobot 51:39–49
Mirski P, Brzosko E (2015) Are hermaphrodites better adapted to the colonization process in trioecious populations of Salix myrsinifolia? Acta Soc Bot Poloniae 84:167–175
Nagamitsu T, Hoshikawa T, Kawahara T, Yu Barkalov V, Sabirov RN (2014) Phylogeography and genetic structure of disjunct Salix arbutifolia populations in Japan. Popul Ecol 56:539–549
Neumann A (1981) Die mitteleuropäischen Salix Arten. Mitteilungen der Forstlichen Bundes-Versuchsanstalt, Wien, p 134
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13:1143–1155
Obbard DJ, Harris SA, Pannell JR (2006) Sexual systems and population genetic structure in an annual plant: testing the metapopulation model. Amer Nat 167:354–366
Pannell JR, Barrett SCH (1998) Baker’s law revisited: reproductive assurance in a metapopulation. Evolution 52:657–668
Pannell JR, Dorken ME (2006) Colonisation as a common denominator in plant metapopulations and range expansions: effects on genetic diversity and sexual systems. Landscape Ecol 21:837–848
Pannell JR, Dorken ME, Pujol B, Berjano R (2008) Gender variation and transitions between sexual systems in Mercurialis annua (Euphorbiaceae). Int J Plant Sci 169:129–139
Peakall ROD, Smouse PE (2006) GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Not 6:288–295
Perdereau AC, Kelleher CT, Douglas GT, Hodkinson TR (2014) High levels of gene flow and genetic diversity in Irish populations of Salix caprea L. inferred from chloroplast and nuclear SSR markers. BMC Plant Biol 14:202. doi:10.1186/s12870-014-0202-x
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distribution. Ecol Model 190:231–259
Pons O, Petit RJ (1996) Measuring and testing genetic differentiation with ordered and unordered alleles. Genetics 144:1237–1245
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pucholt P, Rönnberg-Wästljung AC, Berlin S (2015) Single locus sex determination and female heterogamety in the basket willow (Salix viminalis L.). Heredity 114:575–583
Rambaut A (2010) FIGTREE 1.3.1. Available at: http://tree.bio.ed.ac.uk/software/figtree/ (accessed 10 October 2015)
Rambuda TD, Johnson SD (2004) Breeding systems of invasive plant species in South Africa: does Baker’s rule apply? Diversity Distrib 10:409–416
Ramos-Onsins SE, Rozas J (2002) Statistical properties of new neutrality tests against population growth. Mol Biol Evol 19:2092–2100
Renner SC (2014) The relative and absolute frequencies of angiosperm sexual systems: dioecy, monoecy, gynodioecy, and an updated online database. Am J Bot 101:1588–1596
Richards AJ (1997) Plant breeding systems. Chapman and Hall, London
Sakai AK, Weller SG (1999) Gender and sexual dimorphism in flowering plants: a review of terminology, biogeographic patterns, ecological correlates, and phylogenetic approaches. In: Geber MA, Dawson TE, Delph LF (eds) Sexual and gender dimorphism in flowering plants. Springer-Verlag, Heidelberg, pp 1–31
Skvortsov AK (1968) Willows of the USSR. Nauka, Moscow
Small RL, Ryburn JA, Cronn RC, Seelanan T, Wendel JF (1998) The tortoise and the hare: choosing between noncoding plastome and nuclear Adh sequences for phylogeny reconstruction in a recently diverged plant group. Am J Bot 85:1301–1315
Smulders MJM, Cottrell JE, Lefèvre F, Van Der Schoot J, Arens P, Vosman B et al (2008) Structure of the genetic diversity in black poplar (Populus nigra L.) populations across European river systems: consequences for conservation and restoration. For Ecol Manag 255:1388–1399
Sochor M, Vašut RJ, Bártová E, Majeský L, Mráček J (2013) Can gene flow among populations counteract the habitat loss of extremely fragile biotopes? An example from the population genetic structure in Salix daphnoides. Tree Genet Genomes 9:1193–1205
Stamati K, Hollingsworth PM, Russell J (2007) Patterns of clonal diversity in three species of sub-arctic willow (Salix lanata, Salix lapponum and Salix herbacea). Plant Syst Evol 269(1-2):75–88
Stebbins GL (1984) Polyploidy and the distribution of the arcticalpine flora: new evidence and a new approach. Bot Helv 94:10–13
Steltzer H, Hufbauer RA, Welker JM, Casalis M, Sullivan PF, Chimner R (2008) Frequent sexual reproduction and high intraspecific variation in Salix arctica: implications for a terrestrial feedback to climate change in the high Arctic. J Geophys Res-Biogeo 113:G03S10. doi:10.1029/2007JG000503
Sugier D, Sugier P, Pawełek M, Gawlik-Dziki U (2011) Salix myrsinifolia Salisb. As a source of phenolic glycosides: distribution and characteristic of habitat conditions in the mideastern Poland. Acta Sci Polonorum Hortorum Cultus 10:75–88
Swaegers J, Mergeay J, Therry L, Larmuseau MHD, Bonte D, Stoks R (2013) Rapid range expansion increases genetic differentiation while causing limited reduction in genetic diversity in a damselfly. Heredity 111:422–429
Taberlet P, Gielly L, Patou G, Bouvet J (1991) Universal primers for amplification of three noncoding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Tajima F (1989) Statistical methods to test for nucleotide mutation hypothesis by DNA polymorphism. Genetics 123:585–595
van Dulmen A (2001) Pollination and phenology of flowers in the canopy of two contrasting rain forest types in Amazonia, Colombia. Plant Ecol 153:73–85
van Puyvelde K, Triest L (2007) ISSRs indicate isolation by distance and spatial structuring in Salix alba populations along Alpineupstream rivers (alto Adige and upper Rhine). Belg J Bot 140:100–108
Vamosi JA, Vamosi SN, Barrett SCH (2003) Sex in advertising: dioecy alters the net benefits of attractiveness in Sagittaria latifolia (Alismataceae). Proc R Soc B 273:2401–2407
Vos P, Hogers R, Bleeker M, Reijans M, Vandelee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP—a new technique for DNA-fingerprinting. Nucleic Acids Res 23:4407–4414
Vyskot B, Hobza R (2004) Gender in plants: sex chromosomes are emerging from the fog. Trends Genet 20:432–438
Yeh FC, Yang RC, Boyle T (1999) POPGENE version 1.32: Microsoft Windows-based freeware for population genetic analysis, quick user guide. Center for International Forestry Research, University of Alberta, Edmonton, Alberta, Canada.
Zieliński J (1976) Atlas rozmieszczenia drzew i krzewów w Polsce. PWN, Warszawa-Poznań
Acknowledgments
This paper is part of the research funded by the Polish Ministry of Science and Higher Education (grant no. N304 335439).
Data Archiving Statement
Sequences records are available at GenBank under following accessing numbers:
KU871004, KU871005, KU871006, KU871007, KU871008, KU871009, KU871010, KU871011 and KU871012.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Pär Ingvarsson
Electronic supplementary material
Online Resource 1
Nuclear DNA content determined in three S. myrsinifolia sex morphs: males, females or hermaphrodites (PDF 142 kb)
Online Resource 2
Match distribution of pairwise differences of cpDNA haplotypes for Salix myrsinifolia in north/southern gradient of European range (Lithuania-Poland). Continuous line expected distribution, dotted line observed distribution (PNG 95 kb)
Online Resource 3
Correlation matrix between genetic diversity indices and following variables: N population size, Lat latitude, H R hermaphrodite ratio, F cov ratio of forests in 5.5 km radius, O cov ratio of open natural and half-natural habitats in 5.5 km, h haplotype diversity, π nucleotide diversity, PPL proportion of polymorphic loci, Hj Nei’s gene diversity, I Shannon’s diversity index, DW frequency down-weighed marker value. Statistically significant correlations are given in bold (PDF 146 kb)
Online Resource 4
Number of clusters (ΔK) identified with Bayesian algorithm in STRUCTURE software (PNG 49 kb)
Online Resource 5
Climatic suitability models for Salix myrsinifolia built in MaxEnt software under seven bioclimatic variables (BIO1, BIO2, BIO3, BIO5, BIO7 and BIO12) and averaged from 5 to 8 (depending on period) different global climate models (CCSM, CNRM, FGOALS, GISS, IPSL, MIROC, MPI, MRI). (PNG 869 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Mirski, P., Brzosko, E., Jędrzejczyk, I. et al. Genetic structure of dioecious and trioecious Salix myrsinifolia populations at the border of geographic range. Tree Genetics & Genomes 13, 15 (2017). https://doi.org/10.1007/s11295-016-1096-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-016-1096-6