
Abstract
As part of the Rain In Cumulus over the Ocean Experiment (RICO) and the Puerto Rico Aerosol and Cloud Study (PRACS), cloud water was collected at East Peak (EP) in Puerto Rico. The main objective of this study was to determine the concentrations of water-soluble species (Cl−, NO3 −, SO4 2−, NH4 +, Ca2+, H+, Mg2+, K+, and Na+) in water samples taken from clouds influenced by tropical trade winds. The most abundant inorganic species were Na+ (average 465 µeq l−1) and Cl− (434 µeq l−1), followed by Mg2+ (105 µeq l−1), SO4 2− (61 µeq l−1), and NO3 − (25 µeq l−1). High concentrations of nss–SO4 2 (28 µeq l−1), NO3 − (86 µeq l−1), and H+ (14.5 µeq l−1) were measured with a shift in air masses origin from the North Atlantic to North American continent, which reflected a strong anthropogenic influence on cloud chemistry at EP. Long-range transport of particles and acid gases seems to be the factor responsible for fluctuations in concentrations and pH of cloud water at East Peak. When under trade wind influences the liquid phase concentrations of all inorganic substances were similar to those found in clouds in other clean maritime environments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Cloud water chemical composition and aerosol nucleation scavenging have important consequences for the cloud-processing of different atmospheric aerosols. Those include the removal rate for different species, which affects their atmospheric residence time and thus their influence on indirect and direct aerosol climate forcing. The dissolved constituents of cloud drops are derived either from soluble species contained in aerosol particles that nucleate droplets or from the dissolution of trace gases within droplets (Fuzzi et al. 2002). The different species contained in cloud droplets may then react to form other products that are incorporated into aerosol particles upon droplet evaporation. Clouds and precipitation are also important sources of nutrients and pollutants deposited into aquatic and terrestrial ecosystems (Marinoni et al. 2004). Therefore, the knowledge of cloud and rain chemical composition is also important for evaluating their potential effects on biota.
The role of clouds in the transport, transformation, and delivery of chemical pollutants has recently received increased attention especially in temperate regions (e.g., Choularton 1997; Acker et al. 2002). In tropical latitudes, however, there are few studies examining either the inorganic (Gordon et al. 1994; Clark et al. 1998) or organic (Sanhueza et al. 1992) composition of cloud water. The inorganic components of cloud water have been extensively studied (Falconer and Falconer 1980; Hegg and Hobbs 1981; Collett et al. 1995) primarily because some of these species (e.g., sulfate and nitrate) are important in acid deposition (Khwaja et al. 1995). In temperate regions with strong anthropogenic influence, SO4 2−, NO3 −, H+, and NH4 + often dominate cloud water composition (Weathers et al. 1988; Khwaja et al. 1995; Marinoni et al. 2004; Collett et al. 2002; Brüggemann et al. 2005). Species derived mainly from anthropogenic sources such as H+, NO3 −, and NH4 + have been found at concentrations two to six times lower than those for many temperate locations (Clark et al. 1998). On the other hand, other ionic species such as Na+, Mg2+, Ca2+, and K+ have been shown to be more abundant than in many temperate regions (Clark et al. 1998). Puerto Rico (18°16′ N, 65°45′ W), a tropical island in the Eastern part of the Caribbean, has been the site of few studies related to clouds. Weathers et al. (1988) and Asbury et al. (1994) measured ion concentrations in cloud water and/or rainwater and found high concentrations for Na+ and Cl−, as is consistent with a strong marine influence. Despite the importance of cloud water chemical composition to climate and ecosystem, studies are lacking, mainly in tropical and remote regions. A better knowledge of the relationships between solute concentrations in cloud water and their temporal variations would further improve the construction of cloud chemistry models.
The results presented here are from the Rain In Cumulus over the Ocean experiment (RICO) and the Puerto Rico Aerosol and Clouds Study (PRACS; details are available at http://www.eol.ucar.edu/rico/). The RICO project provided the opportunity to study aerosols (including the organic fraction) and their CCN activity in clouds in a marine tropical environment (e.g., Allan et al. 2008). Here, we present results from cloud water samples collected at a mountain site on Puerto Rico. We aimed to determine the chemical composition of the liquid phase of clouds and to identify possible sources for the water-soluble species.
2 Methodology
2.1 Sampling Site
The sampling site for cloud water collection was located at East Peak (EP) in the El Yunque National Forest, Luquillo, Puerto Rico (see Fig. 1). EP is located in a tropical rainforest (18°16′ N 65°45′ W), 65 km of San Juan, 45 km of the Atlantic Ocean, at an altitude of 1,050 m asl. The area is a humid mountaintop with frequent and abundant cloud coverage fed by trade winds. The sampling period considered here extended from December 2004 through January 2005. Monthly values of rainfall and average temperatures averaged 370 mm and 17°C in December 2004, and 300 mm and 16.7°C in January. The lowest temperature measured in these months was 14.3°C, and the highest was 19.6°C. The wind direction during the study was predominantly NE (from the Atlantic Ocean).

East Peak (EP) sampling site on the island of Puerto Rico. The arrow indicates the predominant wind direction
2.2 Sampling and Analysis of Cloud Water
Cloud water samples were collected using an aluminum version of the compact Caltech Active Strand Cloud water Collector 2 (CASCC-2). This single-stage collector (Al-CASCC2) samples bulk cloud water representative of the entire drop size distribution (Demoz et al. 1996). Samples were collected for each monitored cloud event (1 to 4 h) in eight events. Immediately after the event, the samples were gathered from the collector. An aliquot was used to measure pH and conductivity at the site immediately after collection. Samples were then stored in glass bottles, which have been previously cleaned with Triton 100 x and Milli Q water, and baked overnight at 450°C. The samples then were stored in a freezer at −18°C for further analysis.
The ionic concentrations of cloud water samples were determined using an ion chromatograph (model DIONEX series 4500 for cations and model 500 for anions). The anionic species measured were chloride (Cl−), sulfate (SO4 2−), and nitrate (NO3 −), and the cationic species were ammonium (NH4 +), calcium (Ca2+), magnesium (Mg2+), potassium (K+), and sodium (Na+). A gradient (H2O; NaOH 5 mM; NaOH 100 mM) was used for the analysis of anions with an ION Pac® AS11 column. For the cations, a CS12A column with an eluent of methanesulfonic acid 20 mM was used. The detection limit for each ion was computed as a blank average plus twice the blank standard deviation. For both cations and anions, a calibration check with external standards was performed to assure accuracy of ±5%.
Liquid water content (LWC) was calculated for the January period using the collection rate (ml min−1) that was then converted to LWC (g m−3) according to the approach used by Demoz et al. (1996). They calculated that 2.5 ml min−1 corresponds to 0.5g m−3 using a CASCC2.
Five-day back trajectories (BTs) were calculated for every sampling leg using the hybrid single-particle Lagrangian integrated trajectories (HYSPLIT) computer model (Draxler and Rolph 2003)
2.3 Quality Control and Statistical Analysis of Cloud Water Samples
Field and laboratory blanks were collected and analyzed in the same way as the samples. Between events, the cloud water sampler was cleaned using large amounts of Milli Q water. Once the collector was cleaned, a blank was taken by spraying about 200 ml of Milli Q water onto the collection strands in the collector.
Na+ was used as the reference species for marine origin (Wilson 1975). Non-sea salt (nss) concentrations were calculated for a given species (X) using the following:
where [X] and [Na+] are the concentrations of the species in µeq l−1 in cloud water.
2.4 Cloud Drop Sampling and Analysis of Residual Particles
In December cloud drops of the trade cumuli arriving at EP were sampled by a counterflow virtual impactor (CVI, Ogren et al. 1985). The inlet was originally developed for aircraft use, i.e., it is installed in a wind tunnel for ground-based applications to reach air velocities of about 150 m s−1. A counterflow established at the inlet tip opposite the sampling direction allowed the CVI to presegregate the interstitial particles and gas phase. Only cloud drops with sufficient inertia to overcome the counterflow were sampled. The lower cutoff drop diameter of the CVI system already deployed in other ground-based cloud studies (e.g., Mertes et al. 2005) was 5 µm. The collected drops were injected into particle-free and dry carrier air in the sampling line behind the CVI tip, where they became completely evaporated, releasing dry residual particles, which were attributed to the processed CCN. The drop evaporation in the dry air occurs at room temperature in order to minimize the vaporization of aerosol components in the residuals. The relative humidity in the sampling line is below 5% after the complete collected LWC is driven into the gas phase. Behind the evaporation section, the sample flow is divided into several lines guiding different analyzing devices. The LWC sampled by the CVI was derived from a water vapor measurement using a Lyman-α hygrometer.
The chemical analysis of the residual particles was carried out by an Aerodyne Quadrupole Aerosol Mass Spectrometer (Q-AMS) coupled to the CVI. This instrument measures chemically resolved mass concentrations of various nonrefractory (at 600°C) aerosol compounds, such as sulfate, nitrate, ammonium, and organic matter. Detailed descriptions of the Q-AMS have been published previously (e.g., Jayne et al. 2000; Allan et al. 2003). Briefly, the instrument focuses aerosol particles with vacuum aerodynamic diameters (d va) between 50 and 1,000 nm into a narrow beam by means of an aerodynamic lens. Upon impact on a hot surface (typically 600°C in this study) under high vacuum, the nonrefractory particle components are flash-vaporized. The vapor is subsequently ionized by electron impact (E = 70 eV), and the resultant ions are analyzed by a quadrupole mass spectrometer.
Simultaneously with the Q-AMS, a Scanning Mobility Particle Sizer (SMPS), consisting of a Differential Mobility Analyser (GRIMM model 5.500) and a Condensation Particle Counter (GRIMM model 5.403), measured number size distributions of residual particles with mobility diameters (d mob) between 10 and 520 nm downstream the CVI inlet.
The aerodynamic particle sizing by the Q-AMS and the electrical mobility sizing by the SMPS allow an estimation of the particle density ρ p, since under the assumption of spherical particles, the two quantities d va and d mob are related by d va = (ρ p/ρ 0) d mob where ρ 0 is the unit density (1 g cm−3) (Jayne et al. 2000). From comparison of the two measured size distributions, a density of about 1.7 g cm−3 was inferred. Using this density, the upper detection diameter of the Q-AMS was determined to be a maximum d mob of 590 nm. Thus, atmospheric mass concentrations (µg m−3) of the nonrefractory inorganic ions and organic matter inferred by the Q-AMS referred to cloud drop residual particles having a mobility diameter up to about 590 nm. Liquid phase concentrations (mg l−1 and additionally µeq l−1 for the inorganic ions) of these compounds were derived by dividing mass concentrations by the simultaneously sampled LWC. The aerosol mass found inside the cloud liquid phase was calculated from the SMPS measurements by integration over the residual particle size distribution up to the AMS size limit using the inferred dry particle density of 1.7 g cm−3. Dividing again by the sampled LWC yielded the aerosol mass liquid phase concentration of particles smaller than 590 nm. Thus, the aerosol mass and liquid phase concentration inferred from the SMPS could be directly compared to the AMS results because both instruments measured particles up to the same mobility diameter size range. During the December 2004 period, a Particle Volume Monitor (PVM, Gerber Scientific Inc.) was operated to measure the LWC of the clouds prevailing at East Peak.
3 Results and Discussion
Samples were collected for four events over 2 days in December 2004 and eight cloud water samples over 9 days in January 2005 at East Peak. Here, we compare the cloud chemical composition of the December 2004 and January 2005 using different cloud water samplers. Afterwards, the results of the inorganic cloud water composition will be presented. Finally, probable sources of the measured species are discussed.
3.1 Comparison of Al-CASCC2 and CVI Cloud Observations
Tables 1 and 2 show the liquid phase concentrations of different compounds obtained from the Al-CASCC2 and CVI collections. Unfortunately, no comparisons of simultaneous cloud water collections with Al-CASCC2 and CVI could be presented in this study because no water samples are available for December 2004, and the CVI was not available in January 2005. However, good quantitative agreement between both samplers has been found in midlatitude continental clouds for sulfate and low volatile organic compounds (Brüggemann et al. 2005; van Pinxteren et al. 2005).
Due to the lack of simultaneous cloud sampling, the chemical cloud water composition collected by the CVI and the Al-CASCC2 are only discussed for air masses with similar back trajectories. According to Tables 1 and 2, this was available for clean maritime air masses in the December (CVI) and the first January (Al-CASCC2) period. Liquid phase concentrations of SO4 2− and NO3 − derived from both CVI and Q-AMS were well within the concentration range obtained from the cloud water samples (cf. Tables 1 and 2). Assuming a rather constant cloud water composition for clean maritime air masses at least for the winter period, SO4 2− and NO3 − were mainly associated with CCN in the accumulation mode size range (up to about 600 nm) and were mainly not attached to supermicrometer sea salt particles.
The measured organic mass concentration in the cloud residuals at East Peak during the December measurements were below the detection limit of the Q-AMS, which was 0.5 µg/m3 for organic matter with respect to ambient concentrations (Allan et al. 2008).
NaCl particles are not detectable with the Q-AMS; however, the Q-AMS detected a significant amount of chloride (detected in the Q-AMS as HCl+, m/z 36) in the residuals that was not present in the Q-AMS when sampling interstitial aerosol. Furthermore, a simultaneously operated Q-AMS, located outside the clouds directly at the sea shore upwind of the East Peak (Allan et al. 2008) also did not measure any chloride. Therefore, the chloride detected may have originated from NaCl that reacted with H2SO4 and other acidic sulfates in the cloud droplets, forming HCl. The HCl released to the gas phase may have reacted further with NH3 forming NH4Cl, a certain amount of which may have been scavenged by the cloud droplets. NH4Cl is expected to be detected on m/z 36 because it forms HCl+ under electron impact. The ammonium fragments of NH4Cl were not detected because the detection limit of ammonium in the Q-AMS is higher than that for chloride (DeCarlo et al., 2006). This explanation agrees with the observed ratio of Cl−/Na+, which was lower than in seawater (see above), an indication for chloride loss by conversion to HCl.
In addition, a good agreement is obvious from Table 1 between the sum of mass concentrations of AMS-detectable substances and the aerosol mass concentrations from the SMPS measurements inferred as explained in section 2.4. This closure indicates that most CCN smaller than 590 nm consists of inorganic soluble ions and that no substantial amount of NaCl particles in this size range were present in the droplets.
3.2 Chemical Composition of Cloud Water
Ion concentrations (mg l−1 and µeq l−1) of all species, pH, and LWC (g m−3) for cloud water samples collected at East Peak during January 2005 are presented in Table 2. The most abundant anions were Cl− (average of 434 µeq l−1) and SO4 2− (61 µeq l−1), and the most abundant cations were Na+ (average of 465 µeq l−1) and Mg2+ (105 µeq l−1). Differences between the highest and mean concentrations among cloud water samples varied from three- to fourfold for some ions (NO3 −, and, H+) and about twofold for the others. The measured and calculated conductivity were in agreement with a difference about 10%, which is considered acceptable.
The East Peak ion composition in cloud water sampled with the Al-CASCC2 was similar to sea salt components previously obtained at the same site (Weathers et al. 1988; Asbury et al. 1994). The high concentrations of sea salt (Na+, Cl−) and other ions commonly present in seawater (Mg2+, SO4 2−) observed in the cloud water samples at East Peak were consistent with the proximity of the site to the ocean. On the other hand, previously measured concentrations of ions from anthropogenic (SO4 2−, NH4 +, NO3 −) and mineral (Ca2+) sources were significantly higher than our measurements. Asbury et al. (1994) measured SO4 2− concentrations three times higher, H+ six times, and Ca2+ three times than what were measured in 2005 (Fig. 2). NH4 + concentrations were five times lower in the current study than those measured by Weathers et al. (1988) and Asbury et al. (1994). However, our results were based on a monthly average (January 2005), while previous results were based on an annual average. This factor could explain the differences in concentrations, since temperature, season, photochemistry, and air mass transport patterns change during the year. In the present work, back trajectories were derived, so it is possible to indicate the origin of the air masses and thus the anthropogenic influence. However, in the study of Asbury et al. (1994) at EP, which took place more than a decade ago, no back trajectories were verified (at least was not cited in the paper), therefore, it is not possible to confirm a larger impact of anthropogenic influenced air masses in this study that would explain the differences obvious in Fig. 2.

Comparison of average ion concentrations (µeq l−1) of cloud water collected at East Peak by Asbury et al. (1994) and in this study (2005)
The sources of cloud water solutes in our study were predominantly marine (see Section 3.3), showing concentrations of SO4 2−, NO3 −, and NH4 + much lower than in cloud water from polluted areas in the US, but in the range of those from cloud samples from US coastal sites directly exposed to clean Pacific air masses. Obviously, the differences are also caused by different analytical techniques, sampling period (dry and wet season) and regions (Atlantic Coast, Pacific Coast, inside the continent). At several sites in the eastern US, the concentrations of the main ions measured were SO4 2− > NO3 −, H+ > NH4 +, and pH values ranged from 2.4 to 5.5. In other sites in the western US, the concentrations ranked Cl− > SO4 2− > H+ > Na+ and pH ranged from 3.6 to 5.5 (Weathers et al. 1988). Fog concentrations in San Joaquin Valley, California, US, presented higher concentrations of SO4 2− (two times), NH4 + (245 times), and NO3 − (20 times) than those observed at EP (Collett et al. 1999). Similar to our results, the predominant ions in fogs in a pristine Oregon coast were Na+ and Cl− (Collett et al. 2002; Straub et al. 2007), and similar concentrations were also measured for SO4 2− (60 µeq l−1) and NO3 − (23 µeq l−1; Collett et al. 2002).
3.3 Anthropogenic vs. Marine Sources
Although the predominant wind direction in Puerto Rico is from the northeast (air masses coming from the Atlantic Ocean), continental air masses from the African and the North American continents (carrying African dust and anthropogenic pollution, respectively) may also have influence. African dust arrives predominantly from June to September. During the sampling period, no African dust events reached the island, according to The Total Ozone Mapping Spectrometer aerosol index (TOMs http://toms.gsfc.nasa.gov/aerosols/aerosols_v8.html). The back trajectories indicated that during December 9–10 and January 13–19, air masses arrived mainly from the northeast. However, from the 20th to the 23rd of January, air masses started to come from the North, probably from North America. During this time, some ions of potentially anthropogenic origin (NO3 −, and nss–SO4 2−) had concentrations up to six times higher compared to the period when the air masses were coming from the NE (Fig. 3). The pH also changed during these events (Table 2), with a significant decrease from 6.2 when air masses were coming from the northeast (Jan 13–19) to 4.9 when air masses were coming from North America (Jan 22–23), again suggesting an influence of anthropogenic pollution. Even if concentrations of Na+ and Cl−1 ions were slightly reduced from the clean to the polluted period, the concentrations of ions derived predominantly from seawater (Na+, Cl−1, Mg2+, K+, and Ca2+) did not change significantly between these periods (Fig. 3, Table 2).

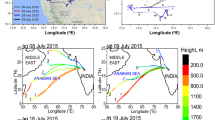
Average percentage (% µeq l−1) of all species found in cloud water at East Peak under different air mass origin with a representative example of back trajectories for both air mass categories
To investigate the marine origin of measured ions, we correlated their concentrations in cloud water. The predominant inorganic species found in all samples were Na+ and Cl−, which are markers for seawater aerosol produced by the sea surface activity of the nearby Atlantic Ocean. Concentrations of Na+ and Cl− were highly correlated (r 2 = 0.94). A lower average Cl−/Na+ ratio (0.93) was found for cloud water than for seawater (1.16) (Table 3). No significant difference in the Cl−/Na+ ratio was observed between periods with and without anthropogenic influence (0.92 and 0.93, respectively). The lower Cl−/Na+ ratio found in cloud water probably reflected some chloride loss from the aerosol due to acid displacement by HNO3 or H2SO4.
Ions usually present in seawater (K+, Mg2+, and Ca2+) correlated well with Na+. The ratios of Mg2+/Na+ K+/Na+, and Ca2+/Na+ were similar to those for seawater, showing that the source for these ions was mainly seawater (Table 3). Nevertheless, low concentrations of nss–Ca2+, nss–Mg2+, and nss–K+ were detected (Table 3). The origin of this non-sea salt component could be soil dust particles from local agricultural or aeolian processes.
The SO4 2−/Na+ ratio (0.2) for cloud water in the polluted period was higher than for the seawater ratio (0.121), suggesting that SO4 2− came primarily, but not exclusively, from sea salt (Table 3). The nss–SO4 2− was 28 µeq l−1, during the influence of air masses from the North America vs. 2 µeq l−1 during the clean period (Table 3).
The NO3 −/Na+ ratios were higher during January 22–23 (avg 0.16) than during January 13–19 (avg 0.03; Table 3). The ratio for these species in seawater is near zero (10–4). Estimating the non-sea salt component yields a concentration of 67 µeq l−1 during the period with anthropogenic influence (January 22–23), and only 11 µeq l−1 was observed in the clean air masses (Table 3). Most of the NO3 − and nss–SO4 2− in cloud water probably came from local sources (natural or anthropogenic) during the period influenced by air masses from the NE (Jan 13–19). The increase in their concentrations during the Jan 22–23 likely reflects long-range transport of polluted air masses from North America (Jickells et al. 1982).
4 Conclusions
Cloud chemical composition at East Peak, Puerto Rico, obtained as part of the RICO project, revealed the influence of marine and continental air masses on cloud chemistry. The most abundant species measured in the cloud water samples were Na+ and Cl−. This is consistent with the proximity of the site to the ocean and suggested that sea salt aerosol is important as CCN. Overall, our results were similar to those found for other remote areas. The history of air masses forming clouds at East Peak had an effect on observed cloud water ion concentrations. When air masses arrived from North America, the SO4 2−/Na+ (0.2) and NO3 −/Na+ (0.16) ratios were much higher than those found in seawater (SO4 2−/Na+~0.12 and NO3 −/Na+ ~0) and cloud pH decreased from 6.1 to 4.9. These differences appeared to reflect long-range transport of NO3 − and SO4 2− derived mainly from emissions of SO2 and NO x in North America. Good agreement between results from both cloud samplers (CVI and Al-CASCC2) for SO4 2− and NO3 − implies that these ions were dominantly associated with CCN of the accumulation mode size range. We also found indications pointing toward conversion of NaCl to HCl by reaction with H2SO4 and other acidic sulfates in cloud droplets.
References
Acker, K., Mertes, S., Möller, D., Wieprecht, W., Auel, R., & Kalaß, D. (2002). Case study of cloud physical and chemical processes in low clouds at Mt. Brocken. Atmospheric Research, 64, 41–51. doi:10.1016/S0169-8095(02)00078-9.
Allan, J. D., Baumgardner, D., Raga, G. B., Mayol-Bracero, O. L., Morales-García, F., García-García, F., et al. (2008). Clouds and aerosols in Puerto Rico—a new evaluation. Atmospheric Chemistry and Physics, 8, 1293–1309.
Allan, J. D., Jimenez, J. L., Coe, H., Bower, K. N., Williams, P. I., & Worsnop, D. R. (2003). Quantitative sampling using an aerodyne aerosol mass spectrometer. Part 1: Techniques of data interpretation and error analysis. Journal of Geophysical Research, 108(D3), 4090. doi:10.1029/2002JD002358.
Asbury, C. E., McDowell, W. H., Trinidad-Pizarro, R., & Berrios, S. (1994). Solute deposition from cloud water to the canopy of a Puerto Rican montane forest. Atmospheric Environment, 28, 1773–1780. doi:10.1016/1352-2310(94)90139-2.
Brüggemann, E., Gnauk, T., Mertes, S., Acker, K., Auel, R., Wieprecht, W., et al. (2005). Schmücke hill cap cloud and valley stations aerosol characterisation during febuko (I): Particle size distribution, mass, and main components. Atmospheric Environment, 39, 4291–4303. doi:10.1016/j.atmosenv.2005.02.013.
Choularton, T. W. (1997). The great dun fell cloud experiment 1993: an overview. Atmospheric Environment, 31, 2393–2405. doi:10.1016/S1352-2310(96)00316-0.
Clark, K. L., Nadkarni, N. M., Schaefer, D., & Gholz, H. L. (1998). Cloud water and precipitation chemistry in a tropical montane forest, Monteverde, Costa Rica. Atmospheric Environment, 32, 1595–1603. doi:10.1016/S1352-2310(97)00384-1.
Collett Jr, J. L., Bator, A., Sherman, D. E., Moore, K. F., Hoag, K. J., Demoz, B. B., Rao, X., & Reilly, J. E. (2002). The chemical composition of fogs and intercepted clouds in the United States. Atmospheric Research, 64, 29–40. doi:10.1016/S0169-8095(02)00077-7.
Collett Jr, J. L., Iovinelli, R., & Demoz, B. (1995). A 3-stage cloud impactor for size-resolved measurement of cloud drop chemistry. Atmospheric Environment, 29, 1145–1154. doi:10.1016/1352-2310(94)00338-L.
Collett Jr, J. L., Hoag, K. J., Sherman, D. E., Bator, A., & Richards, L. W. (1999). Spatial and temporal variations in San Joaquin Valley fog chemistry. Atmospheric Environment, 33, 129–140. doi:10.1016/S1352-2310(98)00136-8.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., et al. (2006). Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Analytical Chemistry, 78, 8281–8289. doi:10.1021/ac061249n.
Demoz, B., Collett Jr, J. L., & Daube Jr., B. C. (1996). On the caltech active strand cloudwater collectors. Atmospheric Research, 41, 47–62. doi:10.1016/0169-8095(95)00044-5.
Draxler, R. R., & Rolph, G. D. (2003). HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory) Model access via NOAA ARL READY Website (http://www.arl.noaa.gov/ready/hysplit4.html). NOAA Air Resources Laboratory, Silver Spring, MD.
Falconer, R. E., & Falconer, P. D. (1980). Determination of cloud water acidity at a mountain observatory in the Adirondack Mountains of New York State. Journal of Geophysical Research, 85, 7465–7470. doi:10.1029/JC085iC12p07465.
Fuzzi, S., Facchini, M. C., Decesari, S., Matta, E., & Mircea, M. (2002). Soluble organic compounds in fog and cloud droplets: what have we learned over the past few years? Atmospheric Research, 64, 89–98. doi:10.1016/S0169-8095(02)00082-0.
Gordon, C. A., Herrera, R., & Hutchinson, T. C. (1994). Studies of fog events at two cloud forests near Caracas, Venezuela—II. Chemistry of fog. Atmospheric Environment, 28, 323–337. doi:10.1016/1352-2310(94)90108-2.
Hegg, D. A., & Hobbs, P. V. (1981). Cloud water chemistry and the production of sulfates in clouds. Atmospheric Environment, 15, 1597–1604. doi:10.1016/0004-6981(81)90144-X.
Jayne, J. T., Leard, D. C., Zhang, X. F., Davidovits, P., Smith, K. A., Kolb, C. E., et al. (2000). Development of an aerosol mass spectrometer for size and composition analysis of submicron particles. Aerosol Science and Technology, 33, 49–70. doi:10.1080/027868200410840.
Jickells, T., Knap, A., Church, T., Galloway, J., & Miller, J. (1982). Acid rain on Bermuda. Nature, 297, 55–57. doi:10.1038/297055a0.
Khwaja, H. A., Brudnoy, S., & Husain, L. (1995). Chemical characterization of three summer cloud episodes at whiteface mountain. Chemosphere, 31, 3357–3381. doi:10.1016/0045-6535(95)00187-D.
Marinoni, A., Laj, P., Sellegri, K., & Mailhot, G. (2004). Cloud chemistry at the Puy de Dome: variability and relationships with environmental factors. Atmospheric Chemistry and Physics, 4, 715–728.
Mertes, S., Lehmann, K., Nowak, A., Maßling, A., & Wiedensohler, A. (2005). Link between aerosol hygroscopic growth and droplet activation observed for hill-capped clouds at connected flow conditions during FEBUKO. Atmospheric Environment, 39, 4247–4256. doi:10.1016/j.atmosenv.2005.02.010.
Ogren, J. A., Heintzenberg, J., & Charlson, R. J. (1985). In-situ sampling of clouds with a droplet to aerosol converter. Geophysical Research Letters, 12, 121–124. doi:10.1029/GL012i003p00121.
Sanhueza, E., Santana, M., & Hermoso, M. (1992). Gas- and aqueous-phase formic and acetic acids at a tropical cloud forest site. Atmospheric Environment, 26, 1421–1426.
Straub, D. J., Lee, T., & Collett Jr., J. L. (2007). Chemical composition of marine stratocumulus clouds over the eastern Pacific Ocean. Journal of Geophysical Research, 112, D04307. doi:10.1029/2006JD007439.
van Pinxteren, D., Plewka, A., Hofmann, D., Müller, K., Kramberger, H., Svrcina, B., et al. (2005). Schmücke hill cap cloud and valley stations aerosol characterisation during febuko (II): organic compounds. Atmospheric Environment, 39, 4305–4320. doi:10.1016/j.atmosenv.2005.02.014.
Weathers, K. C., Likens, G. E., Bormann, F. H., Bicknell, S. H., Bormann, B. T., Daube, B. C., et al. (1988). Cloudwater chemistry from ten sites in North America. Environmental Science & Technology, 22, 1018–1026. doi:10.1021/es00174a004.
Wilson, T. R. S. (1975). Salinity and the major elements of sea water, in Chemical Oceanography, Vol. 1, 2nd ed., edited by J. P. Riley and G. Skirrow, 365–413, Academic, Orlando, Fla.
Acknowledgements
We thank the El Yunque National Forest and the Conservation Trust of Puerto Rico (Cabezas de San Juan natural reserve) for allowing the use of their facilities to collect our samples. We also acknowledge the support of the National Science Foundation (ATM Grant 0342548 and ATM Grant 0222607), the EPSCoR program, and the Max Planck Society (MPG).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gioda, A., Mayol-Bracero, O.L., Morales-García, F. et al. Chemical Composition of Cloud Water in the Puerto Rican Tropical Trade Wind Cumuli. Water Air Soil Pollut 200, 3–14 (2009). https://doi.org/10.1007/s11270-008-9888-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11270-008-9888-4