
Abstract
Infectious bronchitis virus (IBV, genus Gammacoronavirus) causes an economically important and highly contagious disease in chicken. Random primed RNA sequencing was applied to two IBV positive clinical samples and one in ovo-passaged virus. The virome of a cloacal swab pool was dominated by IBV (82% of viral reads) allowing de novo assembly of a GI-13 lineage complete genome with 99.95% nucleotide identity to vaccine strain 793B. In addition, substantial read counts (16% of viral reads) allowed the assembly of a near-complete chicken astrovirus genome, while lower read counts identified the presence of chicken calicivirus and avian leucosis virus. Viral reads in a respiratory/intestinal tissue pool were distributed between IBV (22.53%), Sicinivirus (Picornaviridae, 24%), and avian leucosis virus (37.04%). A complete IBV genome with 99.95% nucleotide identity to vaccine strain H120 (lineage GI-1), as well as a near-complete avian leucosis virus genome and a partial Sicinivirus genome were assembled from the tissue sample data. Lower read counts identified chicken calicivirus, Avibirnavirus (infectious bursal disease virus, assembling to 98.85% of segment A and 69.66% of segment B closely related to D3976/1 from Germany, 2017) and avian orthoreovirus, while three avian orthoavulavirus 1 reads confirmed prior real-time RT-PCR result. IBV sequence variation analysis identified both fixed and minor frequency variations in the tissue sample compared to its in ovo-passaged virus. Metagenomic methods allow the determination of complete coronavirus genomes from clinical chicken samples while providing additional insights in RNA virus sequence diversity and coinfecting viruses potentially contributing to pathogenicity.
Similar content being viewed by others

Introduction
Infectious bronchitis virus (IBV) is a gammacoronavirus of the subgenus Igacovirus (ICTV 2020, [1]) causing an acute, highly contagious upper respiratory tract disease with potential systemic complications in chickens posing a major economic burden on the poultry industry [2]. The virus replicates in the respiratory, digestive, excretory, and reproductive system, resulting in a variety of pathogenic effects and both respiratory and gastrointestinal viral excretions are possible (reviewed in [3, 4]). In addition to respiratory signs, decreased egg quality and production are common. Serological evidence suggests that poultry workers may develop anti-IBV antibodies following exposure to infected birds, although there is no evidence of active infection in humans [5]. The virus is worldwide in distribution, and there are many genetic and antigenic types that can co-circulate [2, 6], complicating control efforts by the available live attenuated and killed vaccines [7, 8]. Moreover, like other Coronaviridae, IBV is under constant evolution resulting from error prone genome copying that accumulates mutations as well as widely documented recombination events [9,10,11]. Laboratory differential diagnostic efforts are essential because of similarities to mild forms of disease caused by agents such as Newcastle disease virus, avian metapneumovirus, infectious laryngotracheitis virus, mycoplasmas, Avibacterium paragallinarum, and Ornithobacterium rhinotracheale and the co-circulation of these pathogens [12].
Coinfections with other poultry pathogens have been described to complicate the infectious bronchitis disease severity. For instance, coinfection with IBV has been documented in H9N2 avian influenza outbreaks in poultry [12] and may contribute to the disease outcome. Coinfecting respiratory pathogens including Mycoplasma gallisepticum, Mycoplasma synoviae, avian influenza virus, IBV, and avian metapneumovirus seem to frequently occur, as described in Algerian poultry flocks [13]. In addition, infectious bronchitis disease course has been documented to be influenced by infectious bursal disease virus and chicken anemia virus induced immunosuppression [12, 14].
Metagenomic methods, here defined as the random sequencing of all nucleic acids in a sample, are increasingly used since the wider availability of Next-Generation Sequencing (NGS) platforms. Potentially detecting all coinfecting pathogens, these methods proved a huge added value in a context where disease outcome is influenced by multiple infection (e.g., [15, 16]). Moreover, the hypothesis-free methodology allows the detection of unexpected pathogens or variants thereof, the investigation of diagnostic cases that have exhausted the available targeted assays (e.g., [17]), and provides a powerful tool for the discovery of novel viruses [18, 19]. Indeed, random metagenomic methods were critical in the identification of SARS-CoV2 as the etiological agent of the current COVID-19 pandemic [20] and novel emerging livestock diseases like Schmallenberg virus [21].
The objectives of this study were (1) to validate metagenomic protocols for sequencing of avian coronavirus complete genomes from clinical samples; (2) the genetic characterization of selected IBV recently circulating in Belgium; and (3) the detection of viruses co-circulating with IBV.
Materials and methods
Samples, pretreatment, and targeted testing
One pooled sample of lung, trachea, and intestinal tissue from five broiler chickens (4439-PTLB) was submitted for Newcastle disease virus (NDV) diagnostics and differential diagnosis and tested weak positive (Cp 36.35) for avian Orthoavulavirus 1 (NDV), as well as Igacovirus (Infectious bronchitis virus of poultry, Cp 32.29) using routine real-time RT-PCR assays [22, 23]. The sample pretreatment included homogenization in sterile PBS (10% weight/volume) with antibiotics (penicillin: 10.106 U/L, streptomycin 10 g/L, gentamycin 0.25 g/L), followed by 10-min centrifugation at 2500 rpm at 4 °C and harvesting of the supernatant. A derived chorioallantoic fluid sample (4439_ECE) was obtained from the later tissue sample following routine virus isolation procedures using two 5-day amplification rounds in the allantoic cavity of 9–11-day-old specific pathogen-free embryonated chicken eggs [24]. In addition, a pool of five chicken cloacal swabs (4134_PCS), in viral transport medium (BHI 37 g/L, supplemented with 107 U/L penicillin, 2 g/L streptomycin, 1 g/L gentamycin, and 650 mg/L kanamycin), testing positive for IBV (Cp 24.82) was included. Both clinical samples were investigated in a context of avian influenza and NDV exclusion diagnostics and differential diagnosis in farms showing increased mortality. The homogenate of the tissue sample pool, the pool of cloacal swabs, and the chorioallantoic fluid resulting from the in ovo passage of the former tissue sample pool were included in this metagenomic study.
Sequencing
The tissue homogenate, swab transport medium, and chorioallantoic fluid were centrifuged for 5 min at 10,000×g in a precooled (4 °C) centrifuge prior to collection of the supernatant. RNA was extracted from the supernatant using a combination of the TRIzol reagent (Thermo Fisher) and RNeasy mini kit (Qiagen), including on-column DNase treatment as previously described [25]. cDNA was synthesized using SuperScript IV reverse transcriptase (Thermo Fisher Scientific) and random hexamer primers, followed by double-strand cDNA synthesis using the NEBNext mRNA second-strand synthesis module (New England BioLabs). Sequencing libraries were prepared using the Nextera XT kit (Illumina) and standard Nextera XT index adapters (Illumina) and sequenced using a MiSeq reagent kit version 3 (Illumina) with 2x300-bp paired-end sequencing aiming for a minimum of 5-million read pairs per sample. Metagenomic NGS data were generated for 3 samples. The resulting fastq raw metagenomics datasets are publicly available in the Sequence Read Archive (SRA) under BioSample accession numbers SAMN19554998 (tissue sample pool), SAMN19554999 (in ovo passage resulting from tissue sample pool), and SAMN19555000 (cloacal swabs pool).
Bioinformatic analysis
Metagenomic read classification: raw NGS reads were trimmed using Trimmomatic v0.38 [26] to remove adapter sequences and low quality bases (setting the ‘ILLUMINACLIP 2:30:10’, ‘SLIDINGWINDOW:4:20’, and ‘MINLEN:50’ options). Only paired reads were retained for further analysis. Classification of trimmed reads was performed with Kraken2 (Galaxy version 2.0.7) as previously described [27]. A customized Kraken database was built using all available RefSeq “Complete Genome” sequences of six targeted taxonomic groups (archaea, bacteria, fungi, human, protozoa, and viral) downloaded from RefSeq Genome (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/) on 18/02/2019. The proportion of reads mapping to the host (Gallus gallus) was determined using a separate database of selected avian genomes (Gallus gallus GCF-000002315; Columba livia GCF-00037935; Meleagris gallopava GCF-000146605; Anas Platyrhynchos GCF-000355855; Numida meleagris GCF-002078875). Read counts per taxonomic level classified by Kraken2 were further normalized as reads per million (RPM) total (trimmed) reads [27] to remove technical bias introduced by sequencing depth variation between samples. An arbitrary acceptance criterion of RPM > 1 for a significant mNGS finding was used as previously described [27].
De novo assembly and complete genome characterization
Raw sequence data was trimmed using Trim Galore! V0.5.0 (q = 25, l = 50, paired; https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The genome of IBV strain IBV/chicken/Belgium/4439_001_iPTLB/2020 was de novo assembled from a random paired subset (2 × 50,000 reads) of the reads resulting from allantoic fluid sample 4439_ECE using SPAdes v3.9.0 [28] and IVA v1.0.0. [29]. Relevant contigs (Igacovirus, as determined using a Blastn search of all contigs longer than 500 nt against the NCBI Nucleotide database) were joined using CAP3 [30]. The full trimmed dataset was mapped to the finished full genome length contig using BWA-0.6.2 [31]. Minimap2 [32], Samtools [33], and bamToFastq (https://bedtools.readthedocs.io/en/latest/content/tools/bamtofastq.html) were used to enrich on-target reads from datasets resulting from the clinical samples, using specific databases for Coronaviridae (NCBI:txid11118, all RefSeq entries on 14/08/2020, n = 66), Sicinivirus (NCBI:txid1755587, all NCBI nt entries with length > 2000 nt, n = 24), avian leucosis virus (NCBI:txid11864, all NCBI nt entries with length > 7000 nt, n = 200), and avian astrovirus (NCBI:txid249589, all NCBI nt entries with length > 7000 nt, n = 64). For data resulting from the cloacal swabs pool 4134_PCS, random paired subsets (2 × 50,000 reads) were used for the de novo assembly of Coronaviridae - and Avastrovirus-enriched reads, respectively, using the aforementioned strategy (IVA, SPAdes, CAP3), followed by mapping of all trimmed reads against the resulting contigs (Minimap2). All Coronaviridae, Sicinivirus, and avian leucosis virus reads enriched from the trimmed datasets resulting from tissue sample pool 4439_PTLB were used for de novo assembly using the aforementioned strategy. For Taxa reported at lower RPM in the Kraken2 analysis, all classified reads were extracted using Minimap2 and the reference genome(s) present in the Kraken2 classification database, followed by de novo assembly using SPAdes v3.9.0 [28] and similarity searches (Blastn) of resulting contigs or singletons.
To identify conserved protein domains, a RPS-BLAST search [34] against the Conserved Domain Database (CDD) was performed [35]. The annotation of assembled genomes and partial genomes was done using GATU [36] relative to references KF377577.1 and MN548287.1 with manual editing of the coronaviral conserved -1 ribosomal frameshift in ORF 1ab for the avian coronavirus genomes.
Identification of sequence variation in Igacovirus RNA population
The Bam files representing the mapping of all quality-trimmed data from the tissue sample 4439-PTLB or 4439_ECE to assembled genome IBV/chicken/Belgium/4439_001_iPTBL/2020 (consensus genome from 4439_ECE) were used to call RNA population frequency variation using Lofreq [37] using filtering settings min_dp_100, sb_fdr (applying a strand bias multiple testing correction using a fdr correction p value > 0.001), min_snvqual_66, and min_indelqual_20. Only variants with a minimum allele frequency of 0.01 and a positional coverage of at least 100 were included.
Phylogenetic analysis
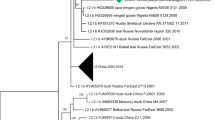
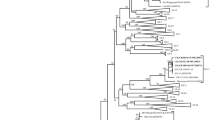
MAFFT v7.310 [38, 39] was used to align the complete S1 coding sequences of infectious bronchitis virus genomes IBV/chicken/Belgium/4439_001-iPTLB/2020 (genome assembled from chorioallantoic fluid 4439_ECE), IBV/chicken/Belgium/4439_001_PTLB/2020 (genome assembled from the tissue sample pool 4439_PTLB), and IBV/chicken/Belgium/4134_001/2019 (genome assembled from the cloacal swab pool, 4134_PCS) with the reference dataset proposed for IBV classification by Valastro and colleagues [6]. The final dataset contained 204 sequences of 1567 nucleotides long. After selection of the most suitable evolutionary model (lowest Bayesian Information Criterion score), a maximum likelihood phylogenetic tree was calculated in Mega X [40] (GTR + G + I [41]; partial deletion of missing data and gaps; 500 bootstrap replicates). For visual clarification, subtrees representing the identified lineages GI-1 and GI-13 were extracted from the full-dataset phylogenetic tree.
Due to low nucleotide sequence similarity with other siciniviruses, the amino acid sequence of the conserved RdRp domain was extracted from all Sicinivirus entries in the NCBI nucleotide database with length > 2000 (n = 15 after removal of duplicates and entries lacking the RdRp domain, NCBI:txid1755587) and aligned with the RdRp region of Sicinivirus strain Belgium/4439_001/2020 and two root sequences representing sister clades (Gallivirus NC018400 and Turdivirus NC014411) using MAFFT v7.310, followed by maximum likelihood phylogenetic analysis using Mega X (LG + G + I model; 500 bootstrap replicates).
The RdRp complete coding sequence as well as the Capsid partial coding sequence of chicken astrovirus strain Belgium/4134/2019 were aligned with 31, respectively, 68 coding sequences available in NCBI nucleotide database using MAFFT v7.310, followed by maximum likelihood phylogenetic analysis using Mega X (LG + G + I model; 100 bootstrap replicates).
Results
Next-generation sequencing raw data
Minimum 5.3 million paired reads were produced per sample (Table 1). As expected for tissues samples, 90% of the reads in sample 4439_PTLB corresponded to the host (chicken), while 1% of reads were viral and minimal bacterial contamination was seen (4%). The results for the corresponding chorioallantoic fluid, 4439_ECE, showed an excellent purity with 77% of the reads being viral and 18% avian, reflecting the viral growth in embryonated chicken eggs. The cloacal swab pool sample 4134-PCS showed a relatively high proportion (10%) of viral reads and as expected for cloacal samples, a high proportion of bacterial reads (56%), and relatively low proportion of host reads (13%).
Virome composition using Kraken2
A diverse virome was detected in pooled tissue sample 4439_PTLB (IBV and NDV RT-PCR positive tissue pooled sample), dominated by IBV (Gammacoronavirus, representing 22.54% of viral reads), Sicinivirus (Picornaviridae, 23.99% of viral reads), and avian leucosis virus (37.04% of viral reads) (Table 2). In addition, lower normalized read counts were detected for chicken astrovirus (0.26% of viral reads), while chicken calicivirus, IBDV (assembling to 98.85% of segment A and 69.66% of segment B of IBDV closely related to D3976/1 from Germany, 2017), and avian orthoreovirus were detected with normalized read counts just above our significance threshold. With only 3 reads detected, avian orthoavulavirus remained below our arbitrary significance level of RPM > 1.
The IBV/chicken/Belgium/4439_001_iPTLB/2020 virus sequenced from the in ovo passage (4439_ECE) of the above sample showed a remarkable increase in the amount and proportion of coronaviral reads (99.80% of viral reads) (Table 2). About half of the avian coronavirus reads are attributed to IBV and half to turkey coronavirus, both being closely related species. Only a single Sicinivirus read was detected in the chorioallantoic fluid sample (possibly carry-over between sequencing libraries), and only low read counts for avian leucosis virus (Retroviridae), while these taxa jointly contributed more than 60% of the viral reads in the clinical sample. Other viruses present in the clinical sample were not detected in its derived chorioallantoic fluid sample. Interestingly, not only gammacoronaviruses were classified by the Kraken2 approach (99.77% of viral reads) but also significant normalized read counts for betacoronaviruses and deltacoronaviruses. However, follow-up investigation (de novo assembly of Kraken2-classified beta- and deltacoronaviral reads followed by Blastn analysis of resulting contigs) showed that these reads mapped to conserved coronaviral sequences. Only gammacoronaviral contigs (IBV H120) were de novo assembled from reads classified by kraken2 as beta- or deltacoronaviral.
82.15% of viral reads from the pool of cloacal swabs 4134_PCS represented avian coronavirus (Gammacoronavirus), while additional alpha-, beta-, and deltacoronaviruses were present at lower normalized read counts (Table 2). About half of the avian coronavirus reads were attributed to IBV and half to turkey coronavirus, both being closely related species. Follow-up investigation (de novo assembly of Kraken2-classified beta- and deltacoronaviral reads followed by Blastn analysis of resulting contigs) showed that non-gamma coronaviral reads mapped to conserved coronaviral sequences. Only gammacoronaviral contigs were de novo assembled from reads classified by kraken2 as beta- or deltacoronaviral. In addition, 16.14% of viral reads represented chicken astrovirus, with some additional astroviral reads from related species. The low number of canine and porcine astrovirus reads represented bioinformatic artifacts (low complexity regions or database biases). Chicken calicivirus and pigeon picornavirus B were detected with significant normalized read numbers. However, follow-up investigation (de novo assembly of Kraken2-classified pigeon picornavirus B reads followed by Blastn analysis of resulting contigs) showed that the pigeon picornavirus B reads mapped to conserved picornaviral RdRp sequences. Avian leucosis virus was detected in the cloacal swab pool above our significance threshold, but with low normalized read numbers as expected because of the low amount of host cells in cloacal swabs.
Low normalized read counts for bacteriophage sequences were observed in the tissue sample and its derived in ovo passage, while a moderate RPM for bacteriophages was detected in the cloacal swab, as well as a few reads classifying as an environmental aquatic virus.
Assembly of viral genomes and molecular characterization
Igacovirus
From the data resulting from the embryonated chicken egg-passaged sample 4439_ECE, a 27,648 bp contig was de novo assembled from Coronaviridae-enriched reads (using an enrichment database of all 66 complete coronavirus genomes in the RefSeq database and extraction of mapped reads). 6 million read pairs aligned to this contig. This complete genome sequence of IBV isolate IBV/chicken/Belgium/4439_001_iPTLB/2020 (submitted to Genbank under accession number MZ367367) has a 99.95% nucleotide identity to IBV strain H120 (MN548287.1), a widely used vaccine of the Mass serotype. Variant calling using Lofreq on the mapped coronaviral reads from the egg-passaged sample (using a minimum positional coverage of 100x) identified two variable genome positions (one in the 5’UTR and one in the envelope protein coding region) with minimum allele frequency of 0.10 as well as 24 positions with minor frequency variation (0.01 ≤ allele frequency ≤ 0.10).
Avian coronavirus reads originating from the clinical tissue sample, 4439_PTLB, from which the above strain was derived in ovo, were mapped to the complete genome sequence from strain IBV/chicken/Belgium/4439_001_iPTLB/2020 (4439_ECE) (47,990 mapped reads), confirming the presence of IBV strain H120-like IBV in the original sample. The resulting genome of IBV/chicken/Belgium/4439_001_PTLB/2020 (submitted under Genbank accession number MZ367368) showed only 1 consensus level mutation difference (in the envelope protein coding region) to the genome from the derived in ovo-passaged sample. Variant calling using Lofreq on the mapped coronaviral reads from the pooled tissue sample and using a minimum positional coverage of 100× as well as a minimum allele frequency of 0.10 indicated the presence of 5 additional genome positions with mixed populations (in the polyprotein 1ab as well as the envelope protein coding regions; Suppl. Table 1). Minor frequency variation (0.01 ≤ allele frequency ≤ 0.10) was detected in 40 additional genome positions scattered throughout the genome (Suppl. Table 1). All variant and minor frequency variant positions detected in the clinical sample were distinct from variant positions detected in the egg-passaged sample (Suppl. Table 1).
A near-complete avian coronavirus genome (27,600 bp contig, submitted under Genbank accession number MZ367369) was de novo assembled from Coronaviridae-enriched reads from the pooled swabs 4134_PCS. About 1.2 million reads aligned to this contig. This near-complete genome sequence obtained from pooled cloacal swabs showed a 99.98% nucleotide identity to IBV strain ck/CH/LLN/130102 (KP118888.1), a 793B genotype strain isolated in China in 2013, and a 99.95% nucleotide identity to IBV vaccine strain 4/91 (KF377577.1, a 793B-type strain).
Phylogenetic classification of the complete S1 coding sequences from the three samples in comparison with 201 reference sequences confirmed the circulation of IBV with close similarity to H120 (4439_PTLB) and 793B B genotype (4134_PCS) vaccine-like strains of lineages GI-1 and GI-13, respectively (Fig. 1), following the classification criteria and nomenclature by Valastro and colleagues [6]. The genomes assembled from the sample 4439_PTLB and the corresponding in ovo-passage IBV/chicken/Belgium/4439_001_iPTLB/2020 from 2020 shared an identical S1 complete coding sequence.

Maximum likelihood phylogenetic analysis of the complete S1 coding sequence of 204 infectious bronchitis virus strains. A GTR + G + I evolutionary model was used with partial deletion of missing data and gaps and 500 bootstrap replicates to evaluate the significance of nodes. For visual clarification, subtrees representing the identified lineages GI-1 and GI-13 were extracted from the full-dataset phylogenetic tree. ♦: sequence determined in the present study. IBV/chicken/Belgium/4439_001_PTLB/2020 and IBV/chicken/Belgium/4439_001_iPTLB/2002 correspond to the genome sequences determined from the tissue sample pool and the derived embryonated egg-passaged virus, respectively, while Infectious bronchitis virus strain IBV/chicken/Belgium/4134_001/2019 corresponds to the sequence determined from the cloacal swabs pool
A partial Sicinivirus genome was de novo assembled from Sicinivirus-enriched reads originating from the pooled tissue sample 4439_PTLB (using an enrichment database containing all 24 Sicinivirus in nt database longer than 2000 nucleotides and extraction of mapped reads). Two contigs were assembled, corresponding to the 5′ end (1347 nt) of MN873047 and the 3′ end (7719 nt) of MN873045, both from chicken fecal samples, USA 2003–2004. These partial sequences were submitted to Genbank under accession numbers MZ367370 and MZ367371. The largest contig encoded the anterior 2573 amino acids of the polyprotein and contains two picornaviral capsid (rhv), an RNA helicase, and an RNA-dependent DNA polymerase (RdDp)-conserved domain. The maximum likelihood amino acid phylogenetic analysis of RdDp conserved region of 28 Sicinivirus sequences (Supplementary Fig. 1) confirmed grouping with other Sicinivirus genomes and lack of geographical clustering which may indicate widespread global circulation of Sicinivirus.
The near-complete genome of an endogenous avian leukosis genome (8532 nt contig, Genbank accession number MZ367376) was de novo assembled from avian leucosis virus-enriched reads from the tissue sample pool 4439_PTLB (enrichment database containing all nt entries longer than 7000 nt). The 8532 nt contig showed 99.95% nucleotide identity to avian leukosis virus genome ev21 (KY235336.1), an avian leukosis subgroup E endogenous virus sequenced from a chicken in China in 2015.
A partial genome (covering positions 8-6478 and 6714-7497, Genbank accession number MZ367372) of a chicken astrovirus was de novo assembled from avian astrovirus-enriched reads originating from the pooled cloacal swabs 4134_PCS (enrichment database containing all 64 nt Avastrovirus entries longer than 7000 in nt database). The partial genome showed 91.96% nt identity (based on longest contig, i.e., position 8-6478) to chicken astrovirus isolate GA2011 (F414802.1) isolated from a chicken in the USA in 2007. Phylogenetic analysis of the Capsid and RdRp complete coding sequences identified strain Belgium/4134_001/2019 as a serotype B chicken astrovirus (Supplementary Fig. 2).
Although only a limited normalized read count was reported, two contigs were de novo assembled from Infectious bursal disease virus-classified reads originating from tissue sample pool 4439-PTLB corresponding to 98.85% of segment A and 69.66% of segment B of an IBDV closely related to recombinant strain D3976/1 from Germany, 2017 (98.63% identity to MN786767.1 segment A and 99.29% identity to MN786769.1 segment B). These partial genome sequences of IBDV/Belgium/4439_001/2020 were submitted to Genbank under accession numbers MZ367373 and MZ367374.
The 908 calicivirus reads identified in the cloacal swab pool were dispersed unequally over the reference (NC033081) genome, resulting in a total coverage breadth of 65.76% of the genome. However, the unequal distribution of reads across the genome only allowed a single contig > 1000 bp to be de novo assembled (1169 nt, coverage 52.99x, Genbank accession number MZ367375) showing a 89.56% nucleotide identity to the partial polyprotein and VP2 coding sequence of a calicivirus from Korean poultry in 2013 (KM254171.1).
Discussion
Since 2006, an enhanced surveillance has been put in place in the Belgian poultry sector for avian influenza. Therapeutic treatment of poultry may only be initiated after the following findings have been submitted to the regional laboratories for examination: a reduction of more than 20% in normal feed and water consumption, a mortality of more than 3% per week, a reduction in egg laying of more than 5% for more than 2 days, or clinical signs or post-mortem lesions suggestive of influenza. Samples analyzed in this study originated from this surveillance and the investigated flocks did not show any specific clinical signs besides increased mortality rates. In addition, vaccination against NDV is compulsory in Belgium and live vaccines are used at young age. Moreover, vaccination against IBV and IBDV is also widely practiced in parallel using live vaccines harboring various residual pathogenicity. In this context, random metagenomic methods provide an important addition to the diagnostic toolbox available to virology laboratories as they provide hypothesis-free identification of nucleic acids of all viruses present in clinical samples [19]. This does not only allow the detection of novel or unexpected viruses, but provides important insights in contexts where multiple infections can complicate the disease manifestation.
Indeed, in addition to providing complete or near-complete Igacovirus genome sequences from IBV-positive clinical samples, the present study documented the presence of additional poultry viruses in clinical samples. This includes the documentation of near-complete genomes of chicken astrovirus from a pooled cloacal swab and Sicinivirus and avian leucosis virus in a pooled tissue sample. Additional poultry viruses were detected with significant normalized read counts including chicken calicivirus, infectious bursal disease virus (IBDV), and avian reovirus. Interestingly, a weak positive real-time RT-PCR positive result for NDV was confirmed with 3 metagenomic reads in a tissue sample pool. Being below our arbitrary significance threshold [27] of RPM > 1, a verification was initiated. Further inspection of these reads identified 3 separate hits to the L gene of MN609929 (NDV/chicken/Vietnam/HU10-1130/2018), ruling out bioinformatic or sequencing artifacts. It was previously stressed that the sensitivity of metagenomic virus detection is a complex interplay between the sample matrix nucleic acid content, the virus genome structure and virus load and that low viral read numbers should not in principle be ruled out as metagenomic findings [42]. However, with only 3 NDV sequence reads, the obtained genetic information does not allow us to hypothesize on the presence of NDV vaccine strain traces vs. circulating field viruses. Of note, the poultry flock received a booster spray vaccination with a NDV live-attenuated vaccine 4 weeks prior to obtaining sample 4439_PTLB.
The comparative metagenomic sequencing of a clinical tissue sample (4439_PTLB) and its derived embryonated chicken egg-passaged sample (4439_ECE) demonstrates a remarkable loss of viral taxa (from the families Picornaviridae, Astroviridae, Caliciviridae, Birnaviridae, and Reoviridae) during routine virus isolation procedures. These viruses are either not easily cultivated or need different biological systems from embryonated chicken eggs routinely used to grow notifiable poultry viruses.
Besides high numbers of gammacoronaviral sequences corresponding to Igacovirus, our original Kraken2 classification identified alpha-, beta-, and deltacoronavirus reads in two samples. However, careful follow-up investigation (extraction of reads classified as alpha beta delta + de novo assembly + blastn similarity searches against the entire nt database) showed that these reads and their resulting contigs aligned to conserved regions of IBV strain H120 genome and hence do not allow a classification below the Coronaviridae family level. Similarly, reads classifying as pigeon picornavirus mapped to a conserved picornaviral sequence (RdRp motif) and thus most likely reflect RdRp sequences from Sicinivirus. The validation of the NDV detection based on only 3 reads and the invalidation of relatively high normalized “non-gamma” coronaviral read counts as conserved coronaviral domains illustrate the critical importance of expert interpretation and follow-up of metagenomic results as was also highlighted in a recent proficiency study [43].
The sequenced Igacovirus genomes from 2019 (pool of cloacal swabs) and 2020 (respiratory and intestinal tissue sample pool) confirm the detection of IBV vaccine strains in Belgium including 4/91 (lineage GI-13) and H120 (lineage GI-1), respectively. However, the limited sampling size does not allow any conclusions about potential circulation of other field and vaccine strains. IBV control relies on a combination of routine vaccination and biosecurity measures [44]. Vaccination programs are complicated by the high genetic variability of the avian coronaviruses, powered both by mutation and recombination events, reducing cross-protection conferred by vaccines toward new variants [45]. Widespread circulation of IBV vaccine strains has been previously documented [46]. A recent detailed survey based on partial S1 gene sequences of IBV strains circulating in Poland suggested that vaccine spreading and persistence seem to occur commonly, stressing the need to further study the epidemiological consequences of the extensive use of live vaccines [46]. In Belgium, a recent study indicated the circulation of at least 7 IBV types, predominated by 4/91-793B and QX-D388, and that a large proportion of the detected IB viruses were vaccine strains [47], confirming a previous study documenting high proportions of vaccine strains as well as a predominant circulation of QX-like field virus [48]. In addition, the genome of a nephropathogenic IBV reference strain isolated in Belgium in 1984 was recently fully sequenced [49].
In Belgium, both H120 (Mass-type) and 4/91 (793B-type) strain-based live-attenuated vaccines are routinely used in poultry [50] and their use in combination is known as “protectotype” approach [8]. Both sampled farms were vaccinated with live-attenuated IBV vaccines according to common practices in Belgium. Unfortunately, the vaccination records of the affected flocks were not able to allow the identification of the exact vaccine used, nor whether the detection of high IBV vaccine strain genome loads in the clinical samples was linked to a recent vaccination event or, alternatively, was a demonstration of circulation or persistence of live-attenuated vaccines. A recent survey in vaccinated poultry in Poland identified one of the applied vaccines in 46.3% of IBV-positive samples [46], while extended persistence of vaccine IBV has been documented previously, especially for 793B vaccines [51, 52].
Comparing the full genome resulting from tissue sample pool 4439_PTLB and 4439_ECE, the corresponding chorioallantoic liquid sample obtained through inoculation, and a subsequent passage in embryonated chicken eggs, only a single consensus level mutation was observed in the envelope small membrane protein coding sequence. This is within the expected range, as previous studies comparing full genome sequences of wild type and egg-adapted IBV focused on high passage attenuation histories (typically 75–100 passages) and documented up to 44 consensus level mutations [53, 54], with mutation hot spots in the polyprotein [53], spike [53, 54], nucleocapsid, and 3’UTR regions [54]. The full IBV genomes were sequenced to a sufficient depth to document virus RNA population differences between clinical sample 4439_PTLB and the corresponding chorioallantoic liquid sample 4439_ECE. Remarkable, a single variant from the clinical sample got fixed in the ECE sample, while all other variants (minimum allele frequency 0.10) and minor frequency variants present in the clinical sample got lost from the RNA population. Moreover, the egg passage introduced 2 new variant sites and 24 new minor frequency variants in the RNA population of 4439_ECE. This indicates a loss of viral diversity from the original population in combination with the generation of novel variation introduced by the passage in embryonated chicken eggs.
An important complementary value of metagenomic methods to targeted diagnostic assays is demonstrated here by documenting the full virome of coinfecting-known poultry viruses, including their high-resolution molecular characterization. In the present study this resulted in (near)-complete genome sequences of a chicken astrovirus of serotype B, an endogenous avian leucosis virus of subtype E, and a Sicinivirus.
Chicken astrovirus is an enteric poultry virus that was identified only in 2004 [55] as a separate species of avian astrovirus. It has been documented globally as an enteric virus in poultry with studies from Poland [56], Jordan [57], India [58, 59], Nigeria [60], China [61], Korea [62], and Brazil [63].
Although its contribution to pathology is often unclear, chicken astrovirus (CAstV) has been associated with poor growth of broiler flocks, enteritis, and diarrhea and is a candidate pathogen in cases of runting stunting syndrome. More recently chicken astrovirus has been implicated in cases of two other diseases of broilers as the sole etiological agent, namely severe kidney disease of young broilers with visceral gout and the “White Chicks” hatchery disease [64]. Our phylogenetic analysis of the Capsid and RdRp coding sequences classifies this first Belgian near-complete chicken astrovirus genome as a member of the predominant serotype CAstV-B [65]. We identified a high normalized astroviral read count in a pool of cloacal swabs, suggesting a high viral load. However, different methodological biases prevent a correlation of normalized read counts and viral load, as some virus families may be preferentially sequenced or identified in bioinformatical workflows.
The present metagenomics study further confirmed the circulation of two recently discovered uncultivable intestinal poultry viruses in Belgium. Sicinivirus was discovered as a new picornavirus with unknown clinical manifestation of poultry in 2012 in Ireland [66] and currently has a worldwide documented distribution [67,68,69,70], while a retrospective metagenomic analysis showed its presence in samples dating back to 2003 [71]. Our study identified a significant normalized Sicinivirus read count in a pool of respiratory and intestinal tissue samples, suggesting a relatively high viral load.
Chicken calicivirus was discovered as an intestinal virus on chicken farms in Germany and in the Netherlands in 2012 [72] and has been confirmed in Brazil, Korea, and the USA (unpublished sequence information cf. NCBI nt database txid1172196 including retrospective identification in a USA sample from 2003). Our study identified a significant but relatively low normalized calicivirus read count in a pool of cloacal swabs, suggesting low viral load.
The identified avian leucosis virus in the tissue sample pool belongs to subgroup E, typically representing endogenous viruses integrated in the host genome [73]. The high normalized read counts for this endogenous virus reflect the high host genomic content (90% of trimmed reads) of the pooled tissue sample. Although only reported with a moderate normalized read count, the Avibirnavirus reads detected in the tissue sample pool assembled to a near-complete segment A and 77% of segment B of infectious bursal disease virus, confirming the previously documented circulation in Europe of recombinant strains with a segment A from very virulent and segment B from attenuated IBDV strains [74]. This finding may be of particular relevance to the observed high IBV genome loads, as IBV disease course can be influenced by IBDV-induced immunosuppression [12, 14].
A limited number of reads (RPM = 3.8) classified as avian orthoreovirus, a globally distributed avian virus, of which some strains cause poultry disease inducing lameness, gastrointestinal, and respiratory and neurological signs, making it relevant in differential diagnosis for notifiable poultry diseases such as avian influenza and Newcastle disease. The reads are mostly distributed along the L3 segment of the single avian orthoreovirus genome included in our Kraken classification database and failed to assemble in meaningful contigs de novo (applying a minimum coverage threshold of 5× and a minimum contig length of 700) [75].
In conclusion, in addition to providing complete genome sequences of viruses present with sufficient viral load in clinical samples—here exemplified by avian gammacoronaviruses-, metagenomic methods allow the detection and characterization of co-circulating viruses that may potentially contribute to the severity of the disease. Moreover, they may facilitate differential diagnosis as exemplified here by the detection of IB, IBDV, NDV, Sicinivirus, Orthoreovirus, and calicivirus in samples originally submitted to the diagnostic laboratory for NDV exclusion. In this sense, these open scope genome detection methods are an important addition to targeted molecular diagnostic assays to unravel clinical issues in livestock. However, a careful follow-up and interpretation of metagenomics findings are essential and reproduction of the disease in controlled conditions might be required.
Availability of data and material
Raw NGS metagenomic data are publicly available in the Sequence Read Archive (SRA) under BioSample accession numbers SAMN19554998-SAMN19555000. Viral genome sequences and partial genome sequences are available under Genbank accession numbers MZ367367-MZ367376.
Code availability
Not applicable.
References
Walker PJ, Siddell SG, Lefkowitz EJ et al (2020) Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch Virol 165:2737–2748
Miłek J, Blicharz-Domańska K (2018) Coronaviruses in avian species—review with focus on epidemiology and diagnosis in wild birds. J Vet Res 62:249–255
Najimudeen MS, Hassan HMS, Cork CS et al (2020) Infectious bronchitis coronavirus infection in chickens: multiple system disease with immune suppression. Pathogens 9:779
Jackwood M, de Wit JJ (2013) Infectious bronchitis. In: Diseases of poultry, 13th edn. Blackwell Publishing Professional, Ames, pp 117–135
Pedersden KA, Sadasiv EC, Chang PW et al (1990) Detection of antibody to avian viruses in human populations. Epidemiol Infect 104:519–525
Valastro V, Holmes EC, Britton P et al (2016) S1 gene-based phylogeny of infectious bronchitis virus: an attempt to harmonize virus classification. Infect Genet Evol 39:349–364
Tizard IR (2020) Vaccination against coronaviruses in domestic animals. Vaccine 38:5123–5130
Jordan B (2017) Vaccination against infectious bronchitis virus: a continuous challenge. Vet Microbiol 206:137–143
Bali K, Bálint Á, Farsang A et al (2021) Recombination events shape the genomic evolution of infectious bronchitis virus in Europe. Viruses 13:535
Hassan MSH, Ojkic D, Coffin CS et al (2019) Delmarva (DMV/1639) infectious bronchitis virus (IBV) variants isolated in Eastern Canada show evidence of recombination. Viruses 11:1054
Jackwood MW, Hall D, Handel A (2012) Molecular evolution and emergence of avian gammacoronaviruses. Infect Genet Evol 12:1305–1311
Bande F, Arshad SS, Omar AR et al (2016) Pathogenesis and diagnostic approaches of avian infectious bronchitis. Adv Virol 2016:4621659
Sid H, Benachour K, Rautenschlein S (2015) Co-infection with multiple respiratory pathogens contributes to increased mortality rates in algerian poultry flocks. Avian Dis 59:440–446
Gallardo RA, van Santen VL, Toro H (2012) Effects of chicken anaemia virus and infectious bursal disease virus-induced immunodeficiency on infectious bronchitis virus replication and genotypic drift. Avian Pathol 41:451–458
Li C-X, Li W, Zhou J et al (2020) High resolution metagenomic characterization of complex infectomes in paediatric acute respiratory infection. Sci Rep 10:1–11
Blomström A-L, Fossum C, Wallgren P et al (2016) Viral metagenomic analysis displays the co-infection situation in healthy and PMWS affected pigs. PLoS ONE 11:e0166863
Moore NE, Wang J, Hewitt J et al (2015) Metagenomic analysis of viruses in feces from unsolved outbreaks of gastroenteritis in humans. J Clin Microbiol 53:15–21
Mokili JL, Rohwer F, Dutilh BE (2012) Metagenomics and future perspectives in virus discovery. Curr Opin Virol 2:63–77
Kwok KTT, Nieuwenhuijse DF, Phan MVT et al (2020) Virus metagenomics in farm animals: a systematic review. Viruses 12:107
Zhu N, Zhang D, Wang W et al (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. https://doi.org/10.1056/nejmoa2001017
Hoffmann B, Scheuch M, Höper D et al (2012) Novel orthobunyavirus in Cattle, Europe, 2011. Emerg Infect Dis 18:469–472
Callison SA, Hilt DA, Boynton TO et al (2006) Development and evaluation of a real-time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens. J Virol Methods 138:60–65
Wise MG, Suarez DL, Seal BS et al (2004) Development of a real-time reverse-transcription PCR for detection of newcastle disease virus RNA in clinical samples. J Clin Microbiol 42:329–338
World Organisation of Animal Health (OIE) (2019) Chapter 3.3.14. Newcastle disease (Infection with Newcastle Disease Virus), manual of diagnostic tests and vaccines for terrestrial animals (terrestrial manual), p 964
Wylezich C, Papa A, Beer M et al (2018) A versatile sample processing workflow for metagenomic pathogen detection. Sci Rep 8:13108
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Van Borm S, Fu Q, Winand R et al (2020) Evaluation of a commercial exogenous internal process control for diagnostic RNA virus metagenomics from different animal clinical samples. J Virol Methods (In Press)
Bankevich A, Nurk S, Antipov D et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Hunt M, Gall A, Ong SH et al (2015) IVA: accurate de novo assembly of RNA virus genomes. Bioinformatics 31:2374–2376
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9:868–877
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li H (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Marchler-Bauer A, Zheng C, Chitsaz F et al (2013) CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 41:D348-352
Oude Munnink BB, Phan MVT, VIZIONS Consortium et al (2017) Characterization of Posa and Posa-like virus genomes in fecal samples from humans, pigs, rats, and bats collected from a single location in Vietnam. Virus Evol 3:vex022
Tcherepanov V, Ehlers A, Upton C (2006) Genome Annotation Transfer Utility (GATU): rapid annotation of viral genomes using a closely related reference genome. BMC Genom 7:150
Wilm A, Aw PPK, Bertrand D et al (2012) LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40:11189–11201
Katoh K, Misawa K, Kuma K et al (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kumar S, Stecher G, Li M et al (2018) MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 35:1547–1549
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
Ebinger A, Fischer S, Höper D (2021) A theoretical and generalized approach for the assessment of the sample-specific limit of detection for clinical metagenomics. Comput Struct Biotechnol J 19:732–742
Höper D, Grützke J, Brinkmann A et al (2020) Proficiency testing of metagenomics-based detection of food-borne pathogens using a complex artificial sequencing dataset. Front Microbiol 11:575377
de Wit JJ, Cook JKA, van der Heijden HMJF (2011) Infectious bronchitis virus variants: a review of the history, current situation and control measures. Avian Pathol 40:223–235
Bande F, Arshad SS, Bejo MH et al (2015) Progress and challenges toward the development of vaccines against avian infectious bronchitis. J Immunol Res 2015:424860
Legnardi M, Franzo G, Koutoulis KC et al (2019) Vaccine or field strains: the jigsaw pattern of infectious bronchitis virus molecular epidemiology in Poland. Poult Sci 98:6388–6392
De Herdt P, De Gussem M, Van Gorp M et al (2016) Infectious bronchitis virus infections of chickens in Belgium:an epidemiological survey. Vlaams Diergeneeskundig Tijdschrift 85:285–290
Worthington KJ, Currie RJW, Jones RC (2008) A reverse transcriptase-polymerase chain reaction survey of infectious bronchitis virus genotypes in Western Europe from 2002 to 2006. Avian Pathol 37:247–257
Reddy VRAP, Theuns S, Roukaerts IDM et al (2015) Genetic characterization of the belgian nephropathogenic infectious bronchitis virus (NIBV) reference strain B1648. Viruses 7:4488–4506
Belgisch Centrum voor Farmacotherapeutische Informatie (BCFIvet) Infectieuze Bronchitis. https://www.vetcompendium.be/nl/node/3448
Cavanagh D, Mawditt K, Britton P et al (1999) Longitudinal field studies of infectious bronchitis virus and avian pneumovirus in broilers using type-specific polymerase chain reactions. Avian Pathol 28:593–605
Tucciarone CM, Franzo G, Berto G et al (2018) Evaluation of 793/B-like and Mass-like vaccine strain kinetics in experimental and field conditions by real-time RT-PCR quantification. Poult Sci 97:303–312
Tsai C-T, Wu H-Y, Wang C-H (2020) Genetic sequence changes related to the attenuation of avian infectious bronchitis virus strain TW2575/98. Virus Genes 56:369–379
Oade MS, Keep S, Freimanis GL et al (2019) Attenuation of infectious bronchitis virus in eggs results in different patterns of genomic variation across multiple replicates. J Virol 93:e00492-e519
Baxendale W, Mebatsion T (2004) The isolation and characterisation of astroviruses from chickens. Avian Pathol 33:364–370
Sajewicz-Krukowska J, Domanska-Blicharz K (2016) Nearly full-length genome sequence of a novel astrovirus isolated from chickens with ‘white chicks’ condition. Arch Virol 161:2581–2587
Lobani AM, Gharaibeh SM, Al-Majali AM (2016) Relationship between different enteric viral infections and the occurrence of diarrhea in broiler flocks in Jordan. Poult Sci 95:1257–1261
Patel AK, Pandit RJ, Thakkar JR et al (2017) Complete genome sequence analysis of chicken astrovirus isolate from India. Vet Res Commun 41:67–75
Panigrahi S, Jindal N, Kumar P et al (2019) Molecular characterization of chicken astroviruses in gout-affected commercial broiler chickens in Haryana, India. Virusdisease 30:551–561
Adebiyi AI, Tregaskis PL, Oluwayelu DO et al (2019) Investigation of enteric viruses associated with runting and stunting in day-old chicks and older broilers in Southwest Nigeria. Front Vet Sci 6:239
Zhao W, Wu Z, Yao Y et al (2020) The isolation and molecular characterization of an astrovirus from “Yellow” Chickens, China. Front Vet Sci 7:822
Koo BS, Lee HR, Jeon EO et al (2013) Molecular survey of enteric viruses in commercial chicken farms in Korea with a history of enteritis. Poult Sci 92:2876–2885
Nuñez LFN, Santander Parra SH, Carranza C et al (2016) Detection and molecular characterization of chicken astrovirus associated with chicks that have an unusual condition known as “white chicks” in Brazil. Poult Sci 95:1262–1270
Smyth VJ (2017) A review of the strain diversity and pathogenesis of chicken astrovirus. Viruses 9:29
Donato C, Vijaykrishna D (2017) The broad host range and genetic diversity of mammalian and avian astroviruses. Viruses 9:102
Bullman S, Kearney K, O’Mahony M et al (2014) Identification and genetic characterization of a novel picornavirus from chickens. J Gen Virol 95:1094–1103
Zhou H, Zhu S, Quan R et al (2015) Identification and genome characterization of the first sicinivirus isolate from chickens in mainland china by using viral metagenomics. PLoS ONE 10:e0139668
Lima DA, Cibulski SP, Finkler F et al (2017) Faecal virome of healthy chickens reveals a large diversity of the eukaryote viral community, including novel circular ssDNA viruses. J Gen Virol 98:690–703
Lima DA, Cibulski SP, Tochetto C et al (2019) The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res 261:9–20
Boros Á, Pankovics P, Adonyi Á et al (2016) A diarrheic chicken simultaneously co-infected with multiple picornaviruses: complete genome analysis of avian picornaviruses representing up to six genera. Virology 489:63–74
Goraichuk IV, Davis JF, Parris DJ et al (2021) Near-complete genome sequences of five siciniviruses from North America. Microbiol Resour Announc 10:e00364-21
Wolf S, Reetz J, Hoffmann K et al (2012) Discovery and genetic characterization of novel caliciviruses in German and Dutch poultry. Arch Virol 157:1499–1507
Fadly AM, Nair V (2008) Leukosis/sarcoma group. In: Diseases of poultry. Iowa State University Press, Ames, pp 514–568
Mató T, Tatár-Kis T, Felföldi B et al (2020) Occurrence and spread of a reassortant very virulent genotype of infectious bursal disease virus with altered VP2 amino acid profile and pathogenicity in some European countries. Vet Microbiol 245:108663
Bányai K, Dandár E, Dorsey KM et al (2011) The genomic constellation of a novel avian orthoreovirus strain associated with runting-stunting syndrome in broilers. Virus Genes 42:82–89
Acknowledgements
This work was supported by funding from the European Union’s Horizon 2020 Research and Innovation programme under Grant Agreement No 773830: One Health European Joint Programme as internal joint research project METASTAVA. Raw sequencing data were professionally generated by the VIB Neuromics Support Facility (Antwerp, Belgium). The authors thank Lotte Weckx for excellent technical assistance and Kevin Vanneste and Raf Winand (Transversal activities in Applied Genomics unit, Sciensano) for providing bioinformatic methods. Dr. Mia Vanrobaeys (DGZ Vlaanderen) and Prof. Ann Garmyn (Ghent University, Faculty of Veterinary Medicine) provided information about the diagnostic cases.
Funding
This work was supported by funding from the European Union’s Horizon 2020 Research and Innovation programme under grant agreement No 773830: One Health European Joint Programme as internal joint research project METASTAVA.
Author information
Authors and Affiliations
Contributions
SVB, FV, EM, and MS performed experiments; SVB, EM, and FV performed data analysis; SVB, MS, TVDB, and BL designed the experiment and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
Not applicable.
Consent for publication
All contributing authors have read and approved of the final version of the manuscript.
Research involving human participants and/or animals
As only bio-banked clinical animal specimens were used, no approval from the ethical committee for animal experiments was needed. Laboratory work and data analysis at Sciensano were not subjected to ethical approval.
Additional information
Edited by William Dundon.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
11262_2021_1872_MOESM1_ESM.pdf
Supplementary file1 (PDF 48 kb) Supplementary Figure 1: Maximum likelihood phylogenetic analysis of the RdRP amino acid sequences of Sicinivirus strains available in public sequence databases. The evolutionary history was inferred using the Maximum Likelihood method based on the Le_Gascuel_2008 model. The tree with the highest log likelihood (-4082,48) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.9025)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 0.00% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 18 amino acid sequences. There were a total of 463 positions in the final dataset. Evolutionary analyses were conducted in in Mega X [40]. The tree was rooted to the branch leading to the Turdivirus and Gallivirus sister clades of Sicinivirus. ♦ : sequence determined in the present study
11262_2021_1872_MOESM2_ESM.pdf
Supplementary file2 (PDF 104 kb) Supplementary Figure 2: Maximum Likelihood phylogenetic analysis of the RdRP complete (panel A) and Capsid partial (panel B) coding sequences of chicken astrovirus. ♦: sequence determined in the present study. The evolutionary histories were inferred using the Maximum Likelihood method based on the General Time Reversible model. The tree with the highest log likelihood (RdRP -3438,70; Capsid (-23594,35) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.4945 and 1.1115 for RdRP and Capsid, respectively)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 55.66% and 15.08% sites for RdRP and Capsid, respectively). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analyses involved 32 (RdRP) and 69 (Capsid) nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. All positions with less than 95% site coverage were eliminated. That is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position. There were a total of 677 (RdRP) and 2032 (Capsid) positions in the final dataset. Evolutionary analyses were conducted in Mega X [40].
11262_2021_1872_MOESM3_ESM.xlsx
Supplementary file3 (XLSX 21 kb) Supplementary Table 1: RNA sequence variants observed in NGS data from clinical sample 4439_PTLB and the derived embryonated egg-passaged virus 4439_ECE. NGS reads were aligned to the consensus the genome assembled from 4439_ECE and sequence variants were reported using Lofreq. Variants with an allele frequency (AF) > 0.10 are highlighted in yellow. An allele frequency of 1.00 signifies a fixed mutation (=100% of reads) in the genome from the clinical sample compared to the corresponding in ovo-passaged sample. In addition to standard quality and strand bias filters in Lofreq, a threshold of a minimum positional coverage of 100 and a minimum allele frequency of 0.01 were used
Rights and permissions
About this article

Cite this article
Van Borm, S., Steensels, M., Mathijs, E. et al. Metagenomic sequencing determines complete infectious bronchitis virus (avian Gammacoronavirus) vaccine strain genomes and associated viromes in chicken clinical samples. Virus Genes 57, 529–540 (2021). https://doi.org/10.1007/s11262-021-01872-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-021-01872-7