
Abstract
We developed and evaluated a reverse transcription loop-mediated isothermal amplification (RT-LAMP) assay for detecting Duck hepatitis A virus type 1 (DHAV-1). The amplification could be finished in 1 h under isothermal conditions at 63 °C by employing a set of four primers targeting the 2C gene of DHAV-1. The RT-LAMP assay showed higher sensitivity than the RT-PCR with a detection limit of 0.1 ELD50 0.1 ml−1 of DHAV-1. The RT-LAMP assay was highly specific; no cross-reactivity was observed from the samples of other related viruses, bacteria, allantoic fluid of normal chicken embryos, or the livers of uninfected ducks. Thirty clinical samples were subjected to detection by RT-LAMP, RT-PCR, and virus isolation, which obtained completely consistent, positive results. As a simple, rapid, and accurate detection method, this RT-LAMP assay has important potential applications in the clinical diagnosis of DHAV-1.
Similar content being viewed by others


Introduction
Duck hepatitis virus type 1 (DHV-1), a member of family Picornaviridae and genus Avihepatovirus, is a kind of single-stranded RNA virus that causes an acute, highly lethal disease in young ducklings called duck hepatitis. Duck hepatitis leads to severe economic losses for duck raising farms. Duck hepatitis virus includes three serotypes DHV-1, DHV-2, and DHV-3. DHV-1 is distributed widely, while DHV-2 and DHV-3 have only been reported in the UK and the USA, respectively [1–7]. Duck hepatitis caused by DHV-1 can lead to mortality up to 95 % in young ducklings during the first week of life, thus accurate and efficient diagnosis is extremely useful to control the initial disease outbreak [1]. Recently, DHV-1 was renamed to DHAV, and DHAV has three genotypes (DHAV-1, 2 ,and 3) [8–11]. DHAV-1 is distributed widely and prevalent in China, DHAV-2 have been only isolated in Taiwan until now [9, 10], while DHAV-3 was first isolated in South Korea [11], but now it is also epidemic in mainland of China.
The traditional detection methods, including virus isolation and neutralization tests, are generally reliable for the diagnosis of DHV-1 [12], but these methods have shortcomings, such as labor intensive, time consuming, and have insufficient sensitivity which cannot detect extremely low viral loads. To address this, a virus antigen-based ELISA was first established in 1991 [13], then a recombinant VP1 protein-based ELISA was developed, which showed agreement with the neutralization test [14]. Nucleic acid-based assays such as RT-PCR, real-time RT-PCR, and real-time quantitative PCR were developed and showed high specificity and sensitivity [1, 8, 15–17]. However, these assays need specialized and expensive equipment such as a thermal cycler or real-time PCR system, thus they are of limited application in rural areas.
Loop-mediated isothermal amplification (LAMP) assay was developed in 2000 [18], which is a novel nucleic acid amplification method that occurs under isothermal conditions. This method employs a DNA polymerase and a set of four specially designed primers that recognize a total of six distinct sequences on the target DNA, which can be amplified with high specificity. LAMP continues with the accumulation of 109 copies of target in less than an hour [18]. As a simple and efficient diagnostic technique, LAMP has been used in the detection of various RNA or DNA viruses, such as Avian leukosis virus [19, 20], Barley yellow dwarf virus [21], swine transmissible gastroenteritis coronavirus [22], avian influenza virus [23], and Foot-and-mouth disease virus [24]. Here, we report a one-step, single-tube RT-LAMP assay for the rapid detection DHAV-1, and its specificity and sensitivity were assessed. This method has potential applications in the early diagnosis and forecasting of DHAV-1.
Materials and methods
Preparation of viruses and bacteria
The DHAV-1 strain (DHAV-SD, Stored at the China General Microbiological Culture Collection Center, CGMCC No.3746) was propagated in the allantoic cavities of 9-day-old SPF chicken embryos. The embryos that died 36–72 h post inoculation were collected. Allantoic fluid was centrifuged (1,000×g at 4 °C for 10 min) and the suspension was stored at −80 °C until it was used for RNA extraction [1].
Duck enteritis virus (DEV), muscovy parvovirus (MPV), avian influenza virus (AIV, H9N2), Riemerella anatipestifer (RA), Salmonella enteritidis, and Escherichia coli (O78), which were maintained in our laboratory, were propagated and the nucleic acids were extracted [1, 25–28].
RNA extraction
Total RNA was extracted from allantoic fluid and liver samples using TRIzol reagent (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions. DHAV-1 total RNA concentration was measured spectrophotometrically at A260 and A280. This RNA was stored at −80 °C before use.
Design of the primers for the RT-LAMP assay
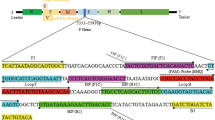
The primers for the RT-LAMP amplification of DHAV-1 were designed based on the conserved region in the 2C gene (GenBank accession no. JX183548). Primers F3, B3, FIP, and BIP were designed by means of the primer software Primer Explorer V4 (http://primerexplorer.jp/elamp4.0.0/index.html; Eiken Chemical Co., Japan). The primer sequences are shown in Table 1.
RT-LAMP
The RT-LAMP reaction was carried out in a total 25 μl reaction volume containing 1 × ThermoPol Reaction Buffer, 8U of Bst DNA polymerase, 10U AMV reverse transcriptase (New England Biolabs, MA, USA), 1 mM dNTP Mix (NewPep, Beijing, China), 0.8 M Betaine, 6 mM MgSO4, 0.2 μM of each of the F3 and B3 primers, 0.8 μM of each of the BIP and FIP primers, and 1.0 μl of the target RNA. The mixture was incubated at 63 °C for 1 h followed by 5 min at 80 °C. After the reaction, the amplified DNA products were detected by electrophoresis on a 1.5 % agarose gel (Biowest Agarose, Spain) followed by ethidium bromide staining under ultraviolet light [21].
RT-PCR
In order to compare the sensitivity of the RT-LAMP assay with other conventional assays, an RT-PCR assay was developed using two pairs of primers (For and Rev; F3 and B3) according to the early report with some changes (Table 1) [8]. The RT-PCR was carried out in a 25 μl total reaction volume using the One-Step RT-PCR kit (NewPep, Beijing, China) with 0.2 μM of each of the upstream and downstream primers and 1 μl of target RNA, according to the supplier’s instructions. The reaction conditions were 42 °C for 30 min, 94 °C for 3 min, 35 cycles at 94 °C for 30 s, 50 °C for 30 s and 72 °C for 20 s, and a final elongation at 72 °C for 10 min. After amplification, the RT-PCR products (250 bp or 213 bp) were analyzed by electrophoresis on an ethidium bromide-stained 1.5 % agarose gel.
Sensitivity comparison of RT-LAMP to RT-PCR
To detect the limit of the RT-LAMP and RT-PCR assay, DHAV-1 total RNAs were extracted from the serially 10-fold diluted allantoic fluid, ranging from 104 to 10−3 50 % egg lethal dose (ELD50) per 100 μl. This single dilution series was used as a template for the two assays. The products were detected by agarose gel electrophoresis as described above (1.5 % agarose, TAE) [21].
Specificity of the RT-LAMP
To assess the specificity of RT-LAMP, including potential cross-reactions with DHAV-1, DEV, MPV, AIV, R. anatipestifer (RA), S. enteritidis, and E. coli (O78) were examined. Total RNA from the allantoic fluid of normal chicken embryos and livers of uninfected ducks were also assayed.
Clinical specimen evaluation
To evaluate the reliability of the RT-LAMP assay, 30 clinical liver samples were collected from DHAV-suspected Ducks in different Provinces of China, including Shandong, Hebei, Sichun, and Beijing. RNA was extracted from these samples and detected by both the RT-LAMP and RT-PCR. The products were detected by agarose gel electrophoresis (1.5 % agarose, TAE). The virus isolation method was also applied to the 30 clinical liver samples using the method previously described [8].
Results
Design of the primers for the RT-LAMP assay
In order to obtain more specificity and detect multiple strains of DHAV-1, the RT-LAMP primers were designed based on a highly conserved region of the 2C gene of the DHAV-1 strain. The one-step, single-tube, RT-LAMP assay was optimized with the selected primer set by varying the ratio of the concentrations of MgSO4 and dNTP, the reaction temperature, and time.
Sensitivity of RT-LAMP
To compare the sensitivity of the RT-LAMP assay with the conventional RT-PCR, the two assays were used to detect the same RNAs which were extracted from 10-fold serial dilutions (from 104 to 10−3 ELD50 per 100 μl) of allantoic fluid. DHAV-1 total RNA concentrations were also measured spectrophotometrically at A260 and A280. Therefore, the corresponding RNA concentration range is from 2 × 104 pg to 2 × 10−3 pg per assay. The results are shown in Fig. 1. The detection limit of the RT-LAMP assay was 0.1 ELD50 per 100 μl, equivalent to 2 × 10−1 pg DHAV-1 total RNA per reaction, which was 100-fold higher than the RT-PCR assay. In addition, the RT-PCR assay using two pairs of primers have the same sensitivity.

Comparison of detection sensitivity between RT-LAMP and RT-PCR by agarose gel electrophoresis. DHAV-1 RNA was serially diluted in 10-fold increments (10−2 to 10−9). a DHAV-1-specific RT-LAMP reaction. b DHAV-1-specific RT-PCR reaction using For and Rev primers. c DHAV-1-specific RT-PCR reaction using F3 and B3 primers. Lane M, DNA size marker (NP5,000, NewPep, Beijing,China); lanes 1–8, extracted total RNA serially diluted in 10-fold increments (10−2 to 10−9)
Specificity of RT-LAMP
The cross-reactivity of the DHAV-1 RT-LAMP assay was evaluated with RNA from DEV, MPV, AIV, RA, S. enteritidis, E. coli (O78), allantoic fluid of normal chicken embryos, and liver of uninfected duck. All these reactions were negative (Fig. 2).

Specificity of the RT-LAMP assay for DHAV-1 detection agarose gel analysis of different viruses and bacteria cross-reaction in the RT-LAMP assay. Lane M DNA size marker (NP5000, NewPep, Beijing, China); lane 1 positive control (DHAV-1 RNA); lanes 2–7 products of DEV, NDV, MPV, AIV, Salmonella enteritidis and Escherichia coli (O78); lane 8 allantoic fluid of normal chicken embryos; lane 9 liver of uninfected duck
Evaluation of RT-LAMP for clinical specimens
To evaluate the feasibility of RT-LAMP of detecting DHAV-1 in clinical specimens, 30 clinical specimens collected over the past years were assayed by RT-LAMP and RT-PCR. In parallel, virus isolation was also performed. The results showed that 11 of the 30 samples tested contained DHAV-1 by virus isolation, the same 11 clinical specimens were also positive by both RT-LAMP and RT-PCR (Fig. 3). The results of RT-LAMP, RT-PCR, and virus isolation were 100 % correlated.

Agarose gel analysis of clinical samples detected by a RT-LAMP and b RT-PCR Lane M DNA size marker (NP5,000, NewPep, Beijing, China); lane 1 positive control (DHAV-1 RNA); lane 2 negative control (liver of uninfected duck); lanes 3–32 clinical samples obtained from different districts of China
Discussion
Several nucleic acid amplification techniques have been developed for the specific and sensitive detection of DHV-1, including RT-PCR and real-time PCR. However, these assays require considerable operator skills, expensive equipment, and 2–4 h for amplification; thus, the application of these assays is limited in the field. Compared to traditional PCR technology, LAMP has more advantages. First, LAMP is more specific since it requires 4 or 6 primers to identify 6 or 8 specific domains [18, 19], while PCR uses only two primers. Second, LAMP is more sensitive, for the amplification of LAMP is more efficient than PCR. Third, LAMP does not require expensive and complex equipment, instead it can be performed using a water bath or heat block for incubation under isothermal conditions. Finally, LAMP is time saving, the assay can be accomplished within 1 h, whereas the PCR technology typically requires 2–4 h [29]. In addition, the LAMP amplification products can be observed by the naked eye directly, as sometimes a white precipitate of magnesium pyrophosphate form during the reaction [30].
After a comparison of different DHAV-1 subgroup genomes, the conserved domain 2C of the genome was selected as the domain for LAMP primer design and was used for screening a group of primers with good amplification efficiency. The 3D gene has also been used to design primers for detection of DHV-1 in the early reports, which encodes an RNA-dependent RNA polymerase [1, 17, 31]. Given that many viruses have RNA polymerase gene, we prefer to choose 2C gene as a detecting marker. A one-step RT-LAMP assay with high specificity and sensitivity was developed for rapid diagnosis of DHAV-1, which has no cross-reaction with DEV, MPV, AIV, R. anatipestifer (RA), S. enteritidis, and E. coli (O78), suggesting that this technique has high specificity to distinguish among some common avian viruses and bacteria at the nucleic acid level. The RT-LAMP has a detection limit of 0.1 ELD50 per 100 μl, equivalent to 2 × 10−1 pg DHAV-1 total RNA per reaction, which was 100 times more sensitive than the conventional RT-PCR, which suggested that this method is useful for the detection of low levels of DHAV-1 and is also useful for confirming the early stages of DHAV-1 infection when viral titers are relatively low.
The RT-LAMP method was also used to detect DHAV-1 in clinical samples. The results from the RT-LAMP assay were consistent with the RT-PCR and viral isolation methods, further confirming the reliability of the RT-LAMP assay. Considering that DHAV-1 RT-LAMP has many advantages, such as being highly sensitive, simple, specific, less time consuming, and not requiring expensive equipment, it is therefore more suitable for use as a DHAV-1 diagnostic tool in the field or rural areas than other nucleic acid-based assays. In summary, the DHAV-1 RT-LAMP assay we developed could be a potential diagnostic method for use in the surveillance, control, and molecular epidemiological screening of DHAV-1 for using in developing countries.
References
M. Yang, A. Cheng, M. Wang, H. Xing, J. Virol. Methods 153, 55–60 (2008)
P.P. Levine, J. Fabricant, Cornell Vet. 40, 71–86 (1950)
P.R. Woolcock, Y.M. Saif, H.J. Barnes, J.R. Glisson, A.M. Fadly, L.R. Mcdougald, D.E. Swayne, Disease of Poultry 11th edn. (Iowa State University Press, Ames IA, 2003), pp. 343–354
C. Ding, D. Zhang, Virology 361, 9–17 (2007)
F.D. Asplin, Vet. Rec. 77, 1529–1530 (1965)
R.E. Gough, M.S. Collins, E. Borland, L.F. Keymer, Vet. Rec. 114, 279 (1984)
R.E. Gough, E.D. Borland, I.F. Keymer, J.C. Stuart, Avian pathol: J. World Vet. Poult. Assoc. 14, 227–236 (1985)
Y. Fu, M. Pan, X. Wang, Y. Xu, H. Yang, D. Zhang, Vet. Microbiol. 131, 247–257 (2008)
C.H. Tseng, H.J. Tsai, Virus Res. 126, 19–31 (2007)
C.H. Tseng, N.J. Knowles, H.J. Tsai, Virus Res. 123, 190–203 (2007)
M.C. Kim, Y.K. Kwon, S.J. Joh, S.J. Kim, C. Tolf, J.H. Kim, H.W. Sung, A.M. Lindberg, J.H. Kwon, Arch. Virol. 152, 2059–2072 (2007)
J. Hwang, Am. J. Vet. Res. 30, 861–864 (1969)
X.L. Zhao, R.M. Phillips, G.D. Li, A.Q. Zhong, Avian Dis. 35, 778–782 (1991)
M. Liu, T. Zhang, Y. Zhang, F. Meng, X. Li, Z. Hou, X. Feng, X. Kong, J. Virol. Methods 169, 66–69 (2010)
C. Anchun, W. Mingshu, X. Hongyi, Z. Dekang, L. Xinran, C. Haijuen, J. Renyong, Y. Miao, J. Microbiol. Meth. 76, 1–5 (2009)
L. Yu-Jun, Z. Gui-Hong, X. Xiao-Qin, C. Jian-Hong, L. Ming, Agric. Sci. China 07, 1140–1146 (2008)
M.C. Kim, Y.K. Kwon, S.J. Joh, J.H. Kwon, J.H. Kim, S.J. Kim, Avian Dis. 51, 540–545 (2007)
T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino, T. Hase, Nucleic Acids Res. 28, E63 (2000)
Y. Wang, Z. Kang, Y. Gao, L. Qin, L. Chen, Q. Wang, J. Li, H. Gao, X. Qi, H. Lin, X. Wang, J. Virol. Methods 173, 31–36 (2011)
X. Zhang, M. Liao, P. Jiao, K. Luo, H. Zhang, T. Ren, G. Zhang, C. Xu, C. Xin, W. Cao, J. Clin. Microbiol. 48, 2116–2121 (2010)
K. Zhao, Y. Liu, X. Wang, J. Virol. Methods 169, 211–214 (2010)
Q. Chen, J. Li, X.E. Fang, W. Xiong, Virol. J. 7, 206 (2010)
M. Imai, A. Ninomiya, H. Minekawa, T. Notomi, T. Ishizaki, P. Van Tu, N.T. Tien, M. Tashiro, T. Odagiri, J. Virol. Methods 141, 173–180 (2007)
J.P. Dukes, D.P. King, S. Alexandersen, Arch. Virol. 151, 1093–1106 (2006)
Y. Li, B. Huang, X. Ma, J. Wu, F. Li, W. Ai, M. Song, H. Yang, Virology 391, 151–161 (2009)
M. Deng, L. Long, X. Xiao, Z. Wu, F. Zhang, Y. Zhang, X. Zheng, X. Xin, Q. Wang, D. Wu, Vet. Immunol. Immunopathol. 141, 183–189 (2011)
S.X. Deng, A.C. Cheng, M.S. Wang, P. Cao, Avian Dis. 52, 88–93 (2008)
G. Le Gall-Recule, V. Jestin, Mol. Cell. Probes 9, 39–44 (1995)
H.T. Chen, J. Zhang, D.H. Sun, Y.F. Chu, X.P. Cai, X.T. Liu, X.N. Luo, Q. Liu, Y.S. Liu, J. Virol. Methods 149, 264–268 (2008)
Y. Mori, K. Nagamine, N. Tomita, T. Notomi, Biochem. Biophys. Res. Commun. 289, 150–154 (2001)
C. Song, H. Wan, S. Yu, X. Han, X. Qiu, Q. Hu, L. Tan, C. Ding, J. Virol. Methods 182, 76–81 (2012)
Acknowledgments
Financial support was provided by the Special Fund for the Agro-scientific Research in the Public Interest (201003012) and the Nature Science Foundation of China (NSFC 31100644).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yang, L., Li, J., Bi, Y. et al. Development and application of a reverse transcription loop-mediated isothermal amplification method for rapid detection of Duck hepatitis A virus type 1. Virus Genes 45, 585–589 (2012). https://doi.org/10.1007/s11262-012-0798-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-012-0798-6