
Abstract
We present results of quasi-classical trajectory (QCT) calculations of the molecular and dissociative sticking probability of CO on Cu(110), and of \(\text {CH}_4\) on Pt(110)-(2 × 1) respectively. Our QCT calculations make use of reactive force fields (RFF) specifically developed for the systems/processes of interest by fitting of a large set of Density Functional Theory (DFT) total energy data. Through these two selected examples of great importance for a deep understanding of relevant chemical reactions on metal surfaces, we illustrate the potentiality of QCT calculations based on accurate RFFs built on ab initio data.







Similar content being viewed by others

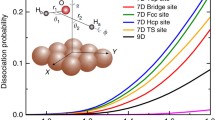
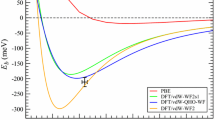
Notes
Quasi-classical is used instead of simply classical, to refer to those trajectory calculations in which the quantized initial roto-vibrational energy of the molecules is taken into account.
References
Hohenberg P, Kohn W (1964) Phys Rev B 136:864
Kohn W, Sham LJ (1965) Phys Rev A 140:1133
Burke K (2012) J Chem Phys 136(15):150901. https://doi.org/10.1063/1.4704546
Kroes GJ, Díaz C (2016) Chem Soc Rev 45:3658. https://doi.org/10.1039/C5CS00336A
Jiang B, Yang M, Xie D, Guo H (2016) Chem Soc Rev 45:3621. https://doi.org/10.1039/C5CS00360A
Maurer RJ, Ruiz VG, Camarillo-Cisneros J, Liu W, Ferri N, Reuter K, Tkatchenko A (2016) Prog Surf Sci 91(2):72. https://doi.org/10.1016/j.progsurf.2016.05.001
Kroes GJ (2012) Phys Chem Chem Phys 14:14966. https://doi.org/10.1039/C2CP42471A
Groß A (2010) ChemPhysChem 11(7):1374. https://doi.org/10.1002/cphc.200900818
Tersoff J (1988) Phys Rev Lett 61:2879. https://doi.org/10.1103/PhysRevLett.61.2879
Brenner DW (1990) Phys Rev B 42:9458. https://doi.org/10.1103/PhysRevB.42.9458
Chenoweth K, van Duin ACT, Goddard WA (2008) J Phys Chem A 112(5):1040. https://doi.org/10.1021/jp709896w
Xiao Y, Dong W, Busnengo HF (2010) J Chem Phys 132(1):014704. https://doi.org/10.1063/1.3265854
Behler J (2016) J Chem Phys 145(17):170901. https://doi.org/10.1063/1.4966192
Kresse G, Hafner J (1994) Phys Rev B 49:14251. https://doi.org/10.1103/PhysRevB.49.14251
Kresse G, Hafner J (1994) J. Phys.: Condens. Matt. 6:8245
Kresse G, Furthmüller J (1996) Phys Rev B 54:11169. https://doi.org/10.1103/PhysRevB.54.11169
Sowa EC, Van Hove MA, Adams DL (1988) Surf Sci 199(1):174. https://doi.org/10.1016/0039-6028(88)90406-2
Korte U, Meyer-Ehmsen G (1992) Surf Sci 271(3):616. https://doi.org/10.1016/0039-6028(92)90923-T
Chadwick H, Gutiérrez-González A, Beck RD, Kroes GJ (2019) J Chem Phys 150(12):124702. https://doi.org/10.1063/1.5081005
Blöchl PE (1994) Phys Rev B 50:17953. https://doi.org/10.1103/PhysRevB.50.17953
Kresse G, Joubert D (1999) Phys Rev B 59:1758. https://doi.org/10.1103/PhysRevB.59.1758
Monkhorst HJ, Pack JD (1976) Phys Rev B 13:5188. https://doi.org/10.1103/PhysRevB.13.5188
Perdew JP, Wang Y (1992) Phys Rev B 45:13244. https://doi.org/10.1103/PhysRevB.45.13244
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865. https://doi.org/10.1103/PhysRevLett.77.3865
Shen XJ, Lozano A, Dong W, Busnengo HF, Yan XH (2014) Phys Rev Lett 112:046101. https://doi.org/10.1103/PhysRevLett.112.046101
Lozano A, Shen X, Moiraghi R, Dong W, Busnengo H (2015) Surf Sci 640:25. https://doi.org/10.1016/j.susc.2015.04.002 Reactivity Concepts at Surfaces: Coupling Theory with Experiment
Wong SSM (1997) Computational methods in physics and engineering. World Scientific, Singapore
Harendt C, Goschnick J, Hirschwald W (1985) Surf Sci 152–153:453. https://doi.org/10.1016/0039-6028(85)90176-1
Anghel AT, Wales DJ, Jenkins SJ, King DA (2005) Phys Rev B 71:113410. https://doi.org/10.1103/PhysRevB.71.113410
Nave S, Tiwari AK, Jackson B (2010) J Chem Phys 132:054705
Sewell TD, Thompson DL (1997) Int J Mod Phys B 11(09):1067. https://doi.org/10.1142/S0217979297000551
Kunat M, Boas C, Becker T, Burghaus U, Wöll C (2001) Surf Sci 474(1–3):114. https://doi.org/10.1016/S0039-6028(00)01041-4
Bonfanti M, Martinazzo R (2016) Int J Quantum Chem 116(21):1575. https://doi.org/10.1002/qua.25192
Bisson R, Sacchi M, Beck RD (2010) J Chem Phys 132(9):094702. https://doi.org/10.1063/1.3328885
Jackson B (213) In: Díez Muiño R, Busnengo HF (eds) Dynamics of gas-surface interactions, Springer Series in Surface Sciences, vol 50. Springer, Berlin, chap 9, pp.213–237. https://doi.org/10.1007/978-3-642-32955-5_9
Acknowledgements
This work was supported by the Consejo National de Investigationes Científicas Técnicas (CONICET) and Ministerio de Educación, Cultura, Ciencia y Tecnología (ME) of Argentina and the Swiss National Science Foundation under the Argentinian-Swiss Joint Research Program (ASJRP) project Nr. IZSAZ2-173328 as well as the ANPCyT project PICT N 2750 (ME-Argentina), and the UNR project PID ING534. Ths authors acknowledge computer time provided by CCT-Rosario Computational Center, and Centro de Simulación Computacional para Aplicaciones Tecnológicas (CSC), members of the High Performance Computing National System (SNCAD, ME-Argentina).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Seminara, G.N., Peludhero, I.F., Dong, W. et al. Molecular Dynamics Study of Molecular and Dissociative Adsorption Using System-Specific Force Fields Based on Ab Initio Calculations: CO/Cu(110) and \(\text {CH}_4/\text {Pt(110)}\). Top Catal 62, 1044–1052 (2019). https://doi.org/10.1007/s11244-019-01196-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-019-01196-9