
Abstract
Reaction of Na2[PdCl4] with two equivalents of amino- or acetylamino-pyridines (LH) affords trans-[PdCl2-(LH)2] {LH = 2-amino-3-methylpyridine (2-ampyH), 3-aminopyridine (3-apyH), 2-acetylamino-3-methylpyridine (2-acmpyH), 3-acetylamino-pyridine (3-acpyH)}. An X-ray crystal structure of trans-[PdCl2(2-ampyH)2] shows that the 2-ampy-H ligands are coordinated in a monodentate fashion via the nitrogen atoms of the pyridine rings. Treatment of trans-[PdCl2(2-acmpyH)2] with NEt3 affords the cyclometalated complex, trans-[Pd(κ2-2-acmpy)2], the X-ray structure of which shows that the 2-acmpy ligand is coordinated to palladium in a bidentate fashion via the nitrogen atom of the pyridine ring and oxygen. Reaction of trans-[PdCl2(LH)2] with two equivalents of sodium saccharinate affords the bis(saccharinate) complexes, trans-[Pd(sac)2(LH)2], in which the saccharinate anions are coordinated via the amide nitrogen atom.
Similar content being viewed by others

Introduction
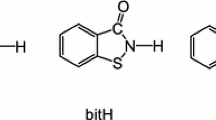
There is continued interest in the chemistry of saccharinate (sac) complexes as co-ligands in biological studies [1, 2], and consequently, a number of reports have recently appeared on the synthesis of platinum and palladium saccharinate complexes [3–11]. While saccharinate can bind to metal centres in a variety of different ways [1, 2] at the relatively soft Pd(II) and Pt(II) centres, it is always N-bound (Fig. 1). Thus, the inclusion of other N-bound ligands to these metal centres results in the formation of an MN4 coordination sphere. Continuing our recent studies in this area [12–14], we herein provide details of the synthesis of four new palladium saccharinate complexes, trans-[Pd(sac)2(LH)2], derived from commercially available or readily prepared amino- or acetylamino-pyridines. Yilmaz and co-workers have also recently detailed the synthesis of related complexes with pyridine and substituted pyridine co-ligands [6–11] and have studied their biological properties [16–19].

Saccharinate (sac) and its most common metal binding mode
Experimental
Materials and methods
All reactions were carried out in the open air using standard bench reagents. 1H and 13C{1H} NMR spectra were recorded on Varian Unity 500 and Gemini 2000 spectrometers, respectively, with CDCl3, d6-dmso or d7-dmf as solvent and internal reference. IR spectra were recorded on a Shimadzu FT-IR 8400 spectrophotometer in the 400–4,000 cm−1 range using KBr discs and in the 200–600 cm−1 using CsI discs. Elemental analyses were carried out at Martin-Luther-Universität. Melting points were measured on a Gallenkhamp melting point apparatus and are uncorrected. Na2[PdCl4], 3-aminopyridine and 2-amino-3-methylpyrimidine were purchased and used as supplied. 3-Acetylamino-pyridine (3-acpyH) [20] and 2-acetylamino-3-methylpyridine (2-acmpyH) [21] were prepared by literature methods. The latter was recrystallized from benzene/hexane (ca 1:1) and characterized prior to use. 2-acmpyH: White prisms, 91 %. Anal Calc. for C8H10N2O: C, 64.0; H, 6.7; N, 18.7. Found: C, 64.0; H, 6.6; N, 18.8; IR (KBr): 3236 s, 3028w, 2956w, 1666 s, 1527 s cm−1. 1H NMR (CDCl3): δ 8.22 (dd, 1H, py), 3 J(HH) = 6.0 Hz, 4 J(HH) = 1.2 Hz, 7.6 (d, 1H, py), 3 J(HH) = 7.5 Hz, 7.12 (dd, 1H, py), 3 J(HH) = 7.5 Hz, 2.28 (s, 3H, CH3), 2.32 (s, 3H, CH3) ppm. Melting point: 52 °C.
Synthesis of 1a–d
A solution of 2-amino-3-methylpyridine (2-ampyH) (0.11 g, 1.0 mmol) in ethanol (10 cm3) was added to a solution of Na2[PdCl4] (0.15 g, 0.50 mmol) in ethanol (10 cm3). The mixture was stirred at room temperature for 3 h. The yellow solid thus formed was filtered off, washed with water and ethanol, and dried under vacuum to give trans-[PdCl2(2-ampyH)2] (1a) (0.16 g, 93 % yield). The related complexes trans-[PdCl2(3-apyH)2] (1b), trans-[PdCl2(2-acmpyH)2] (1c) and trans-[PdCl2(3-acpyH)2] (1d) were prepared and isolated in a similar manner. Crystals of 1a.2dmf suitable for single-crystal diffraction analysis were grown at 25 °C upon standing a dmf solution for several days. 1a: Yellow solid, 93 %. Anal Calc. for C12H16Cl2N4Pd: C, 32.2; H, 4.9; N, 12.5. Found: C, 32.2; H, 4.8; N, 12.3 %; IR (KBr) : 3340 s, 3334 s, 2950w, 1620 s, 1479 s, 1271 m, 1200 m, 751 m, 513 m, 338 m cm−1. 1H NMR (d7-dmf): δ 8.52 (d, 1H, py), 7.55 (d, 1H, py), 7.28 (s, 2H, NH2), 6.73 (t, 1H, py), 2.34 (s, 3H, CH3) ppm, 3 J(HH) = 5.7–7.0 Hz. 1b: Yellow solid, 88 %. Anal Calc. for C10H12Cl2N4Pd: C, 32.8; H, 3.3; N, 15.3. Found: C, 33.1; H, 3.2; N, 15.6 %; IR (KBr): 3445 s, 3337 s, 3020w, 1627 s, 1596 s, 1568 m, 1498 s, 758 m, 461w, 347 s cm−1. 1H NMR (d6-dmso): δ 8.33 (d, 1H, py), 7.43 (t, 1H, py), 7.32 (s, 2H, NH2), 6.57 (d, 1H, py), 6.44 (t, 1H, py) ppm, 3 J(HH) = 8.4 Hz, 4 J(HH) = 5.6 Hz. 1c: Pale yellow solid, 91 %. Anal Calc. for C16H20Cl2N4O2Pd: C, 40.1; H, 4.2; N, 11.7. Found: C, 40.5; H, 4.4; N, 12.0 %; IR (KBr): 3240 s, 3070w, 2977 m, 2927 m, 1689 s, 1596 s, 1598, 792 s, 528 m, 355 m cm−1. 1H NMR (d7-dmf): δ 10.74 (bs, 1H, NH), 9.03 (d, 1H, py), 8.06 (d, 1H, py), 7.55 (dd, 1H, py), 2.26 (s, 3H, CH3), 2.58 (s, 3H, CH3) ppm, 3 J(HH) = 5.4–7.4 Hz. 1d: Yellow solid, 89 %. Anal Calc. for C14H16Cl2N4O2Pd: C, 37.4; H, 3.6; N, 12.5. Found: C, 37.6; H, 3.7; N, 12.7 %; IR (KBr): 3317 s, 3060w, 2877 m, 1701 s, 1578 m, 773 s, 513 m, 364 m cm−1. 1H NMR (d6-dmso): δ 10.4 (s, 1H, NH), 8.28 (d, 1H, py), 8.04 (d, 1H, py), 7.74 (t, 1H, py), 7.06 (t, 1H, py), 2.08 (s, 3H, CH3) ppm, 3 J(HH) = 8.2 Hz, 4 J(HH) = 3.7 Hz.
Synthesis of 2
Several drops of NEt3 were added to a suspension of trans-[PdCl2(2-acmpyH)2] (1c) (0.20 g, 0.44 mmol) in ethanol (10 cm3). The mixture was stirred at room temperature for 3 h. The pale yellow solid was filtered off, washed with ethanol and dried in a vacuum oven (0.12 g, 60 %). Crystals of 2 suitable for single-crystal diffraction analysis were grown by the slow evaporation of a saturated CHCl3 solution. 2: Pale green-yellow solid, 60 %. Anal Calc. for C16H18N4O2Pd : C, 48.8; H, 4.8; N, 13.8. Found: C, 48.5; H, 5.0; N, 14.0 %; IR (KBr): 3097w, 2958w, 1579 s, 767 m, 505w cm−1. 1H NMR (CDCl3): δ 8.26 (dd, 1H, py), 7.44 (dd, 1H, py), 6.72 (dd, 1H, py), 2.29 (s, 3H, CH3), 2.16 (s, 3H, CH3) ppm, 3 J(HH) = 6.1–7.1 Hz, 4 J(HH) = 0.8–1.2 Hz. Melting point: 206–208 °C.
Synthesis of 3a–d
A solution of sodium saccharinate (0.10 g, 0.50 mmol) in ethanol (10 cm3) was added to a suspension of trans-[PdCl2(2-ampyH)2] (1a) (0.10 g, 0.25 mmol) in ethanol (7 cm3). The mixture was stirred for 4 h. The cream-coloured precipitate was filtered off, washed with water and ethanol, then dried under vacuum (0.098 g, 98 % yield). The complexes trans-[Pd(sac)2(3-apyH)2] (3b), trans-[Pd(sac)2(2-acmpyH)2] (3c) and trans-[Pd(sac)2(3-acpyH)2] (3d) were prepared and isolated in a similar manner. 3a: Pale yellow solid, 98 %. Anal Calc. for C26H24N6O6PdS2: C, 45.5; H, 3.5; N, 12.2. Found: C, 45.8; H, 3.7; N, 12.4 %; IR (KBr): 3438 s, 3344 s, 3083w, 2955w, 1652 s, 1596 m, 1307 s, 1257 s, 1166 s, 530w cm−1. 1H NMR (d6-dmso): δ 8.31 (s, 1H, py), 7.83–7.65 (m, 6H, NH2, sac), 7.1 (d, 1H, py), 6.5 (d, 1H, py), 1.98 (s, 3H, CH3). 3b: Pale yellow solid, 91 %. Anal Calc. for C24H18N4O6PdS2: C, 43.7; H, 3.1; N, 12.8. Found: C, 43.8; H, 3.30; N, 13.0 %; IR (KBr): 3427 s, 3311 m, 3209 m, 3088w, 1666 s, 1635 s, 1601 s, 1452, 1254 s, 1163 s, 761 s, 530 s cm−1. 1H NMR (d6-dmso): δ 8.33 (d, 1H, py), 7.86–7.58 (m, 4H, sac), 7.41 (t, 1H, py), 7.27 (s, 2H, NH2), 6.59 (d, 1H, py), 6.50 (t, 1H, py) ppm, 3 J(HH) = 8.0 Hz. 3c: White solid, 80 %. Anal Calc. for C30H28N6O8PdS2: C, 47.3; H, 4.7; N, 9.9. Found: C, 47.1; H, 4.9; N, 10.0 %; IR (KBr): 3240w, 3188w, 2968w, 1703 s, 1652 s, 1595 s, 1251 s, 1164 s, 756 m, 522 m cm−1. 1H NMR (d6-dmso): δ 9.94 (s, 1H, NH), 8.24 (d, 1H, py), 8.05 (d, 1H, sac), 7.87–7.70 (m, 3H, sac), 7.65 (d, 1H, py), 7.18 (dd, 1H, py), 2.14 (s, 3H, CH3), 2.03(s, 3H, CH3) ppm, 3 J(HH) = 8.0 Hz. 3d: Pale yellow, 92 %. Anal Calc. for C28H24N6O8PdS2: C, 45.3; H, 3.3; N, 11.3. Found: C, 45.6; H, 3.4; N, 11.4 %; IR (KBr): 3263 s, 3215w, 3080w, 1714 s, 1655 s, 1581 m, 1437 s, 1254 s, 1165, 761 m, 517 m cm−1. 1H NMR (d6-dmso): δ 10.4 (bs, 1H, NH), 8.29–7.07 (m, 8H, py and sac), 2.10 (s, 3H, CH3) ppm.
X-ray structure determinations
Crystallographic data for 1.2dmf and 2 were collected at 200(2) and 220(2) K, respectively, on a STOE-IPDS diffractometer with Mo-Kα radiation (λ = 0.7103 Å, graphite monochromator). Absorption corrections were made using the IPDS software package [22]. All structures were solved by direct methods with SHELX-97 [23] and refined using full-matrix least-square routines against F2 with SHELXL-97 [24]. Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were included in the models by calculating the positions (riding model) and refined with calculated isotropic displacement parameters. Details of crystallographic data, collection parameters and structure refinement are summarized in Table 1.
Results and discussion
Synthesis of trans-[PdCl2(LH)2] (1a–d) and X-ray crystal structure of trans-[PdCl2(2-ampyH)2] (1a)
In order to prepare the target bis(saccharinate) complexes, trans-[Pd(sac)2(LH)2] (3), we first coordinated a range of amino- and acetylamino-pyridines to the palladium(II) centre. Thus, treatment of Na2[PdCl4] with two equivalents of aminopyridines or acetylamino-pyridines (LH = 2-apyH, 3-ampyH, 2-acmpyH or 3-acpyH) in EtOH afforded trans-[PdCl2(LH)2] (1a–d) in 82–93 % yield as illustrated in Scheme 1. Spectroscopic and analytical data are in full accord with the proposed formulations. In order to ascertain the precise coordination sphere, crystals of 1a.2dmf were grown upon slow evaporation of a saturated dmf solution and the results of an X-ray crystallographic study are summarized in Fig. 2 and its caption. The structure shows the expected square-planar palladium(II) centre ligated by trans pairs of chloride and aminopyridine ligands, and bond lengths and angles are within the expected ranges [25, 26]. The two aminopyridine ligands are related by the inversion centre and thus adopt a relative anti-arrangement with the aryl rings lying approximately perpendicular to the PdCl2N2 plane. The amine substituents do not take part in the bonding to palladium but are in close contact with the dmf solvate [O···H8 2.258 Å] (as shown in Fig. 2), which is also weakly bound to a chloride [Cl···H9 2.712 Å].

Preparation of 1a–d

The molecular structure of trans-[PdCl2(2-ampyH)2] (1a).2dmf with selected bond lengths (Å) and angles (°); Pd(1)–Cl(1) 2.313(1), Pd(1)–Cl(2) 2.279(1), Pd(1)–N(1) 2.016(3), Pd(1)–N(2) 2.023(3), C(1)–N(1) 1.319(5), C(1)–N(3) 1.320(6), C(8)–N(2) 1.307(6), C(8)–N(4) 1.333(6), Cl(1)–Pd(1)–Cl(2) 177.94(4), N(1)–Pd(1)–N(2) 177.6(1), Cl(1)–Pd(1)–N(1) 90.72(9), Cl(2)–Pd(1)–N(1) 88.79(9)
Synthesis and X-ray crystal structure of trans-[Pd(2-acmpy)2] (2)
Addition of a few drops of NEt3 to a suspension of trans-[PdCl2(2-acmpyH)2] (1c) in ethanol afforded trans-[Pd(2-acmpy)2] (2) as a pale yellow solid (60 % yield) (Scheme 2). Crystals of 2 were grown upon slow evaporation of a CHCl3 solution, and the results of an X-ray crystallographic study are summarized in Fig. 3 and its caption. The molecule closely resembles that of the unsubstituted derivative [27, 28] and contains a square-planar palladium(II) centre ligated by two chelating 2-acetylamino-3-methylpyridinate ligands. Palladium–nitrogen and palladium–oxygen bonds are 2.045(2) and 1.973(3) Å, respectively, and the ligand bite angle is 90.29(9)°. The bonding within each of the metallacyclic rings is localized in nature, as seen by the two different carbon–nitrogen bond distances to the nonmetal-bound nitrogen atom [C(1)–N(2) 1.375(4), C(7)–N(2) 1.304(4) Å].

Preparation of 2

The molecular structure of trans-[Pd(2-acmpy)2] (2) (symmetry operation-x + 2, -y,-z) with selected bond lengths (Å) and angles (°); Pd–N(1) 2.045(2), Pd–O 1.973(3), C(1)–N(1) 1.362(4), C(5)–N(1) 1.364(4), C(1)–N(2) 1.375(4), C(7)–N(2) 1.304(4), C(7)–O 1.286(4), O–Pd–O# 180, N(1) –Pd–O 90.29(9), Cl(1) –N(2) –C(7) 125.5(2), Pd–O–C(7) 125.4(2)
Synthesis and characterization of trans-[Pd(sac)2(LH)2] (3a–d)
As stated in the introduction, the aim of this work was to prepare a series of bis(saccharinate) complexes containing a PdN4 core. This was readily achieved by the simple substitution of the chlorides in 1a–d. Thus, treatment of trans-[PdCl2(LH)2] (1a–d) with two equivalents of sodium saccharinate in ethanol afforded trans-[Pd(sac)2(LH)2] (3a–d) in 80–98 % yield (Scheme 3). Characterization was straightforward, being made on the basis of analytical and spectroscopic data. The 1H NMR spectra of each displayed the expected signals for the pyridine or aminopyridine ligands together with a series of multiplets between δ 7.07–8.28 corresponding to the eight protons of the saccharinate ligands. IR spectra showed strong bands within the 1,652–1,666 cm−1 range attributed to ν(CO) of the saccharinate anion. Unfortunately, we have been unable to grow crystals of any of these complexes suitable for single-crystal X-ray diffraction. Nevertheless, on the basis of the NMR data, we can see that a single isomer of each exists in solution. In earlier work, we [13, 15] and others [10] have noted that in some instances rotational isomers (rotamers) are seen in solution. Thus, due to steric requirements, all four nitrogen-based ligands lie perpendicular to the PdN4 plane and these can adopt differing relative positions (syn or anti) of the substituents on sets of like ligands, leading to four possible rotamers of which only two have been observed in the solid-state [13, 15]. That adopted appears to depend upon the development of intramolecular hydrogen bonds between the two different ligand types. In the absence of crystallographic data, we cannot comment further on the isomer that is adopted for 3a–d.

Preparation of 3a–d
Summary and conclusions
This work has further shown that the synthesis of bis(saccharinate) complexes of the type trans-[Pd(sac)2(LH)2] containing amino- or acetylamino-pyridine ligands is general and straightforward starting from Na2[PdCl4] and commercially available ligands. Further, the yields of each step are high, reactions can be carried out without the exclusion of oxygen and water and product isolation and crystallization is simple. Such complexes are known to show biological activity [16–19], while the related phosphine complex trans-[Pd(PPh3)2(sac)2] has been shown to be an efficient catalyst for Suzuki–Miyaura and Negishi cross-coupling reactions [29, 30]. We are currently accessing the biological properties and catalytic properties of 3a–d and related bis(saccharinate) complexes, the results of which will be reported in due course.
Supplementary material
CCDC 999301 and 999302 contain the supplementary crystallographic data for this article. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data-_request/cif.
References
Baran EJ, Yilmaz VT (2006) Coord Chem Rev 250:1980
Baran EJ (2005) Quim Nova 28:1
Henderson W, Nicholson BK, McCaffery LJ (1999) Inorg Chim Acta 285:145
Henderson W, Nicholson BK, Chung DC (2002) Acta Cryst. E58:m432
Santana MD, García-Bueno R, García G, Sánchez G, García J, Kapdi AR, Naik M, Pednekar S, Pérez J, García L, Pérez E, Serrano JL (2012) Dalton Trans 41:3832
Guney E, Yilmaz VT, Kazak C (2010) Polyhedron 29:1285
Guney E, Yilmaz VT, Sengul A, Buyukgunor O (2010) Inorg Chim Acta 363:438
Guney E, Yilmaz VT, Ari F, Buyukgungor O, Ulukaya E (2011) Polyhedron 30:114
Guney E, Yilmaz VT, Buyukgungor O (2010) Inorg Chim Acta 363:2416
Guney E, Yilmaz VT, Buyukgungor O (2011) Polyhedron 30:1968
Yilmaz VT, Ertem A, Guney E, Buyukgungor O, Anorg Z (2010) Allg. Chem. 636:610
Al-Jibori SA, Al-Nassiry AIA, Hogarth G, Salassa L (2013) Inorg Chim Acta 398:46
Al-Jibori SA, Al-Jibori QKA, Schmidt H, Merzweiler K, Wagner C, Hogarth G (2013) Inorg Chim Acta 402:69
Al-Jibori SA, Al-Jibori MHS, Hogarth G (2013) Inorg Chim Acta 398:117
Al-Jibori SA, Habeeb AT, Al-Jibori GHH, Dayaaf NA, Merzweiler K, Wagner C, Schmidt H, Hogarth G (2014) Polyhedron 67:338
Ulukaya E, Ari F, Dimas K, Ikitimur EI, Guney E, Yilmaz VT (2011) Eur J Med Chem 46:4957
Ari F, Ulukaya E, Sarimahut M, Yilmaz VT (2013) Bioorg. Med. Chem. 21:3016
Ari F, Aztopal N, Icsel C, Yilmaz VT, Guney E, Buyukgungor O, Ulukaya E (2013) Bioorg. Med. Chem. 21:6427
Icsel C, Yilmaz VT (2013) DNA Cell Biol 32:165
Hughes MN, Rutt KJ (1972) J Chem Soc Dalton Trans1311
Teotonio ES, Espinda JG, Brito HF, Malta OL, Oliveire SF, Aria DL, Izumi CM (2002) Polyhedron 21:1837
IPDS-Software Package (1999) Stoe and Cie
Sheldrick GM (1997) SHELXS-97. Program for Crystal Structure, Göttingen
Sheldrick GM (1997) SHELXS-97. Program for Refinement of Crystal Structures, Göttingen
Kuduk-Jaworska J, Puszko A, Kubiak M, Pełczyńska M (2004) J Inorg Biochem 98:1447
Qin Z, Jennings MC, Puddephatt RJ (2001) Inorg Chem 40:6220
Scheller-Krattiger V, Scheller KH, Sinn E, Martin BR (1982) Inorg Chim Acta 60:45
Nonoyama M, Tomita S, Yamasaki K (1975) Inorg Chim Acta 12:33
Shah P, Santana MD, García J, Serrano JL, Naik M, Kapdi AR (2013) Tetrahedron 69:1446
Sánchez G, García J, Martínez M, Kapdi AR, García L, Serrano JL (2011) Dalton Trans 40:12676
Acknowledgments
We thank the University of Tikrit for partial support of this work and Erasmus Mundus programme for a postdoctoral scholarship to S.B.-M.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Al-Jibori, S.A., Al-Nassiry, R.A.Q., Al-Jibori, G.H.H. et al. Palladium(II) saccharinate complexes trans-[Pd(sac)2(LH)2] with amino- and acetylamino-pyridine co-ligands: molecular structures of trans-[PdCl2(2-ampyH)2].2dmf (2-ampyH = 2-amino-3-methylpyridine) and trans-[Pd(κ2-2-acmpy)2] (2-acmpyH = 2-acetylamino-3-methylpyridine). Transition Met Chem 39, 735–740 (2014). https://doi.org/10.1007/s11243-014-9845-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-014-9845-6