
Abstract
Various Pd amounts (5 and 20 %wt) were chemically deposited on two different carbon materials (activated carbon and carbon black). Support materials were characterized using low-temperature N2 adsorption and FTIR spectroscopy. SEM images and X-ray diffraction patterns were obtained for the samples tested. Cyclic voltammetric curves in 0.1 M H2SO4 were recorded over a variable sweep potential range for carbon materials with and without Pd. For comparison, the same electrochemical measurements were performed for powdered palladium. The hydrogen electro-oxidation potential decreased and the removal of adsorbed hydrogen by activated carbon-supported palladium was facilitated, which enables these systems to be used as anodes in hydrogen–oxygen fuel cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Palladium supported carbon materials (activated carbon, carbon fibres, carbon black, nanotubes) are highly suitable catalysts for several chemical and electrochemical reactions [1–13]. Relevant concerns regarding the preparation of efficient Pd catalysts include the effects of support pretreatment, reducing agents, surfactants and electrode preparation. Attempts have been made to obtain highly active catalysts in order to achieve optimal efficiencies at low metal loadings. Other work has demonstrated the high activity of carbon-black-supported Pd (promoted with Ru) for hydrogen peroxide electroreduction, which makes this system promising as a cathode catalyst for fuel cells using H2O2 as oxidant [14]. Numerous electrochemical studies of Pd/C systems were performed [2, 3, 5, 8, 10, 12–15] to explore the surface-active states (or sites) of dispersed Pd in various electrolyte solutions. This seems to be the most active electrocatalyst for the anodic oxidation of various organics in low pH solution [16, 17]. Interest in Pd electrochemistry has arisen because of its ability to absorb hydrogen evolved during cathodic polarization. Evolution of H2 and the saturation of the metallic phase by the gas should depend on Pd dispersion and its interaction with the carbon support. Differences in the electrochemical behavior of Pd/C-based electrodes were due to the electronic conductivity and structural properties of the catalysts supported on different carbon materials [5]. The effects of increasing the content of palladium deposited on carbon materials (from 2 to 10%) on the shape of CV curves were studied in acidic solution [2, 18]. At low catalyst loadings, a multiplicity of peaks appears in the hydrogen region in the cathodic and anodic branches. The influence of surface functional groups of the carbon support on palladium dispersion and catalytic activity in hydrogen oxidation was also assessed [19]. However, the literature data relating to the influence of oxygen-containing surface groups on these phenomena are inconsistent [20]. Generally, thermal pre-treatment is often found to be a dominant factor in determining Pd dispersion on a carbon surface.
The objective of this article was to attempt to explain the electrochemical behavior of systems consisting of two kinds of carbon support with various amounts of deposited palladium. The carbon materials used have various surface properties and different porosities. Here, we aim to evaluate the influence of electrode material composition on hydrogen evolution potential, the oxidation of hydrogen and palladium, and the reduction of palladium oxides in acidic electrolyte solution.
Experimental
Materials preparation
Two types of starting carbon materials were used in the experiments: Norit RO 08 activated carbon (AC) and XC-72 (Vulcan) carbon black (CB). AC was previously heat-treated at 1700 °C in a helium atmosphere and then demineralized [21, 22], while CB was used as received. Palladium was deposited by the impregnation of carbon materials as in the method described by Jackson and Shaw [23]. Sufficient palladium(II) chloride solution was added to a quantity of carbon material and fixed at 90 °C for 1 h to thoroughly wet the material and achieve a proper loading (5 and 20 %w/w of Pd deposited on carbon material). Then lithium hydroxide was added and the whole mixture was fixed for a further 3 h. The resulting solid was collected by filtration, then washed with acetic acid solution and finally dried under vacuum at 60 °C for 6 h. The materials were reduced at 200 °C for 1 h under stream of hydrogen. The samples were denoted by unified symbols that specified the kind of carbon used (AC and CB) and the content of Pd (in wt%).
Material characterization
Textural characterization of the tested materials was carried out by N2 adsorption isotherm at 77.4 K. FTIR spectra of the carbon supports were recorded on a FTIR Spectrum 2000 Perkin-Elmer (USA) spectrometer using the KBr pellets technique. Standard pH measurements of a carbon slurry in a neutral electrolyte (0.1 M NaNO3) as well as neutralization of the carbon surface using 0.1 M NaOH and HNO3 were used to evaluate the surface chemistry of the support materials.
Ex situ X-ray diffraction (XRD) and scanning electron microscope (SEM) investigations were carried out to characterize the starting and impregnated materials. The X-ray diffraction patterns were obtained in the 2ϑ = 5–50° range for powdered samples, using the CuKα radiation and HZG-3 powder diffractometer, controlled by IBM PC unit.
Scanning electron microscopy (SEM) images were obtained using a scanning electron microscope SEM 515 made by Philips (Netherlands) under the following conditions: working distance of 14 mm, accelerating voltage of 15 kV and digital image recording DISS.
To make a quantitative check of the Pd content, an XRF analysis of the materials obtained was done using a Canberra Packard-ND-Data apparatus. This analysis showed that desirable amounts of Pd were obtained with an accuracy of nearly to two percent (5 ± 0.1 and 20 ± 0.4%).
Cyclic voltammetry
The cyclic voltammetry measurements were done with an Autolab (Eco Chemie) modular electrochemical system equipped with a PGSTAT 10 potentiostat driven by GPES3 software, and the typical three-electrode electrochemical cell similar to that presented in one of our earlier studies [24]. The working, counter and reference electrodes were a powdered electrode, a Pt wire and a saturated calomel electrode (SCE), in order. The working electrode design is the same as that used and described earlier [24]. After vacuum desorption (10−2 Pa), the powdered sample (mass 100 mg) was placed in an electrode container and drenched with a de-aerated 0.1 M H2SO4 solution to obtain a 3–5 mm sedimentation layer. After assembly, the electrochemical cell was de-aerated until the potentiometric response (E st) of the working electrode had stabilized (20 h). Next, cyclization (scan rate 5 mV s−1) was carried out, and CV curves were recorded when their shape was reproducible (no change in repeated CV scans) [25]. The potentials for hydrogen evolution were determined by independently prepared electrodes. The CV curves were recorded for various potential ranges: the cathodic part between 0.4 V and a different potential in hydrogen region (up to −0.4 V); the anodic part between −0.4 V and a different potential in the oxygen region [26].
Results and discussion
Specific surface areas (S BET), micropore and mesopore volumes (V mi and V me) as well as mesopore surface areas (S me) calculated from nitrogen adsorption isotherms of the carbon samples (Fig. 1) are summarized in Table 1. The surface area of the CB is much larger than that of sample AC. High temperature treatment caused a drastic decrease in the porosity of the starting activated carbon (Norit RO 0.8) [21, 22]. The textural changes in heat-treated activated carbon were studied and discussed earlier for wide temperature range [27]. For temperatures below 1800 °C, the gradual decrease of surface area and microporosity is observed. Above these temperature, the drastic collapse of porosity takes place [27]. On the other hand, thermal treatment near these temperatures can enhance the catalytic activity of the metal/activated carbon system [21, 22]. Both tested materials exhibit a 1.5–2.5 times higher mesopore than micropore volume (in contrast to typical activated carbons). FTIR spectra (Fig. 2) of the carbon materials after subtraction of the KBr pellet spectrum and the arbitrarily selected background (sigmoid line) of the IR absorption edge do not show the presence of active surface oxides. The relatively small bands of structural oxygen (C–O–C) moieties (near 1100 cm−1) and surface OH (3350 cm−1) indicate only slight oxidation of the surface as a result of aeration at ambient temperature. The pH of the carbon slurry (Table 1)—slightly basic (CB) or acidic (AC)—as well as the neutralization results point to the absence of oxygen-containing surface functional groups.

Low-temperature N2 adsorption isotherms for carbon supports (AC, CB) and activated carbon supported palladium systems (AC-5% Pd, AC-20% Pd)

FTIR spectra of carbon supports
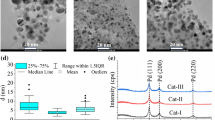
The topography of carbon materials with and without Pd deposits was investigated using scanning electron microscopy. SEM images for all samples (magnification 15 kX) are presented in Fig. 3. It can be seen that the morphology of the starting materials exhibit typical differences—AC (image AC) is a matrix of defected graphite-like microcrystallites, while CB (image CB) shows agglomerates of spheroidally shaped particles. Palladium deposited on the AC-5% Pd surface (image AC-5% Pd) gives isolated metallic clusters of linear size smaller than 1 μm. Increasing the Pd content (image AC-20% Pd) does not enlarge clusters; the entities are still isolated, although they are now located closer to one another. The metal deposited on the CB surface (images CB-5% Pd and CB-20% Pd) creates ramified entities that have grown into the carbon structure. Metal deposition leads to significant changes in the porous structure of the materials obtained. The isotherms of low temperature N2 adsorption on activated carbon supported palladium samples exhibit strongly decreasing values of surface area (S BET), micropore volume (V mi) and mesopore area (S me), see Table 1. The deposition of 20% of palladium almost completely filled the porous structure of the activated carbon support. For carbon black samples, the decrease in these parameters after metal deposition proved to be very much smaller (Table 1).

SEM images of the samples tested
X-ray diffraction patterns for the activated carbon and carbon black samples in selected ranges (2θ between 37–41° and 45–50°) are shown in Fig. 4. In the case of the AC (lines AC, AC-5% Pd, AC-20% Pd), the peaks corresponding to bulk Pd(111) with 2θ near 40.1° and Pd(200) with 2θ near 46.6° (attributed to a face-centered cubic (fcc) structure) [5, 15] are much smaller than those of the CB samples (lines CB, CB-5% Pd, CB-20% Pd) with the same Pd content. For these carbon materials, the height of both peaks rises with increasing Pd-content. In AC samples, the deposited Pd filling up the porous structure of the support probably creates a more dispersed phase, whereas on carbon black, it forms metallic microcrystallites. Generally, XRD data suggest that the average particle size of Pd deposited on CB is larger than that deposited on AC.

XRD patterns of the samples tested
The potentiometric response (Est) in 0.1 M H2SO4 solution for electrodes AC and CB after Pd deposition becomes more positive and closer to the values*obtained with a powdered Pd electrode (Table 2). These values are higher for the AC samples (with and without Pd) than for the CB samples. The differences between the potentials of these materials (with the same Pd content) decreases from 111 mV (samples without Pd) to 36 mV (samples with 20% Pd).
CV curves were recorded for all the electrode materials tested at various cathodic or anodic reverse potentials until the evolution of gaseous hydrogen or oxidation of carbon took place respectively. The anodic and cathodic parts of the CV curves were recorded independently (Figs. 5, 6). The substantial effect of the double layer (C dl increase, see Table 2) was observed; this is not the case with standard electrodes [28, 29]. But the electrode materials studied here seem to resemble the electrode systems used in fuel cells. The hydrogen evolution potentials (Table 2) for electrodes from both support materials and Pd/C systems are more negative than for the Pd powder electrode, but in the case of the CB samples, the values are even more negative. Increasing the Pd content gives less negative potential values. Figs. 5 and 6 show the cathodic parts of the cyclic voltammograms for the AC and CB samples, respectively, without and with deposited Pd recorded from +400 mV (vs. SCE) to various negative reverse potentials (−150, −200, −300, −400 mV). These measurements enable the changes in hydrogen electroreduction and oxidation to be tracked. The gradual increase of reverse potential in activated carbon supported palladium samples causes the successive appearance of double reduction (c) and oxidation (a,a′) peaks (Fig. 5). The curves recorded at the maximal potential range show the peak reduction (ca 270 mV) of the hydrogen ions (in the adsorption layer) and hydrogen evolution wave (hydrogen absorption). As a response, there are two anodic peaks: the first (a), in the range from −220 mV (AC) to −80 mV (AC-20% Pd), corresponds to the oxidation of adsorbed/absorbed hydrogen, while the second one, (a’, only for 20% Pd-containing electrodes) in the potential region from −80 to −20 mV, is ascribed to the oxidation of chemisorbed hydrogen [2, 30]. Peak currents depend strongly on the amount of metal supported on carbon. Higher capacity currents were recorded with electrodes prepared from carbon black (Fig. 6), which may have been due to its larger specific surface area (ca 2.5 times). Only a broad hydrogen oxidation peak (a) appears with Pd-containing electrodes at a potential of −40 mV (CB-5% Pd) or −20 mV (CB-20% Pd). In this case, the peak current depends only slightly on the amount of metal. The cyclic voltammograms recorded for powdered Pd electrodes (Fig. 7) exhibit a broad peak (or two overlapping peaks) in the potential range from 0 to +80 mV. Two possible models of the electro-oxidation of the hydrogen evolved at the cathodes are discussed in the literature [31]. According to the first one, hydrogen dissolved in the subsurface layer of palladium plays the main role during electro-oxidation [31]. In the second one [32], the transition between the α- and β-phases of sorbed electroreduced hydrogen has to be taken into consideration. Thus, one can assume the possible mechanism of electrode processes in the cathodic branch of activated carbon-supported palladium systems to be:

Cyclic voltammograms of activated carbon based electrodes at rising cathodic reverse potentials

Cyclic voltammograms of carbon black based electrodes at rising cathodic reverse potentials

Cyclic voltammograms of a powdered Pd electrode at rising cathodic (solid lines) and anodic (dashed lines) reverse potentials
Reduction
Oxidation
With electrodes prepared from carbon black and powdered palladium electrode, only the electro-oxidation of adsorbed/absorbed hydrogen take place according to reaction (5).
A comparison of the dominant hydrogen electro-oxidation potentials for AC-20% Pd (Fig. 5), CB-20% Pd (Fig. 6) and powdered Pd (Fig. 7) electrodes (−80, −20 and 80 mV, in order) shows that the presence of a carbon (especially activated by thermal pre-treatment) support decreases the oxidation potential of electrochemically generated hydrogen, which energetically facilitates the removal of gaseous hydrogen from the electrode material.
According to literature data [19] and our XRD results on thermally pre-treated activated carbon, a more dispersed metallic palladium phase is formed. Thus, Pd dispersion shifts the hydrogen electrooxidation potential to more positive values. The amount of deposited Pd also influences electrooxidation probably because of the chemisorption of some of the electrochemically evolved hydrogen [2].
In order to evaluate the influence of the electro-oxidation of palladium (and possibly the carbon support) on the electroreduction–oxidation of hydrogen, CV curves were recorded over an extended anodic potential range (up to 700 mV vs. SCE). Figs. 8, 9 and 7 (dashed lines) show the anodic parts of the voltammograms for activated carbon and carbon black-supported Pd as well as powdered Pd, respectively. No electro-oxidation is observed with the AC electrode (curves AC, Fig. 8), but the presence of Pd gives rise to oxidation peaks (a) and an overlapping double reduction peak (curves AC-5% Pd and AC-20% Pd, Fig. 8). Carbon black is not electrochemically oxidized (curves CB, Fig. 9), but the presence of Pd gives only an indistinct anodic wave and cathodic response (curves CB-5% Pd and CB-20% Pd, Fig. 9). In comparison, the powdered Pd electrode (Fig. 7) behaves in much the same way as described in the literature [2, 3, 10, 15, 33]. There is a Pd/Pd2+ couple with an anodic peak potential of ca 600 mV vs. SCE and a cathodic peak potential of ca 350 mV vs. SCE with all the carbon electrodes tested. The double cathodic peak present on the CVs of activated carbon-supported palladium systems indicates the presence of various forms of palladium oxides on the activated carbon surface [34, 35]. Palladium electro-oxidation does not affect the oxidation or reduction of hydrogen during cathodic polarization for AC (see Figs. 5, 8) and CB-based electrodes (see Figs. 6, 9). During both cathodic and anodic polarization (curves 3 in Figs. 5, 8) the AC-20% Pd electrode behaves in the same way as a powdered Pd electrode (Fig. 7). This confirms our earlier suspicion that in this sample, the surface of carbon support accessible to an electrolyte is almost totally covered by the metallic phase (Table 1).

Cyclic voltammograms of activated carbon based electrodes at rising anodic reverse potentials

Cyclic voltammograms of carbon black based electrodes at rising anodic reverse potentials
Conclusions
Electrochemical studies indicate that activated carbon (specially prepared) used as a support for dispersed palladium lowers the hydrogen electrooxidation potential of electrochemically generated hydrogen, which makes it energetically easier to remove adsorbed hydrogen from electrode materials. This diminished potential affects the electromotive force of the hydrogen fuel cell. Moreover, broadening the potential range in the anodic direction does not disrupt the electrochemical activity of the electrode. These facts are important as regarding the selection of carbon materials to be used as supports for active metallic phases in anodic systems in hydrogen–oxygen fuel cells.
References
Albers P, Burmeister R, Seibold K, Prescher G, Parker SF, Ross DK (1999) J Catal 181:145
Pattabiraman R (1997) Appl Catal A Gen 153:9
Andonoglou PhP, Jannakoudakis AD (1997) Electrochim Acta 42:1905
Lupu D, Biriş AR, Mişan I, Jianu A, Holzhüter G, Brukel E (2004) Int J Hydrogen Energy 29:97
Yang W, Yang S, Guo J, Sun G, Xin Q (2007) Carbon 45:397
Villa A, Wang D, Dimitratos N, Su D, Trevisan V, Prati L (2010) Catal Today 150:8
Guo XF, Kim YS, Kim GJ (2010) Catal Today 150:22
Lin Y, Cui X, Ye X (2005) Electrochem Commun 7:267
Bonarowska M, Burda B, Juszczyk W, Pielaszek J, Kowalczyk Z, Karpiński Z (2001) Appl Catal B 35:13
Yang CC, Kumar AS, Zen JM (2006) Electroanalysis 18:64
Grigoriev SA, Fateev VN, Middieton H, Saetre TO, Millet P (2008) Int J Nuclear Hydrogen Prod Appl 1:354
Zheng JS, Zhang XS, Li P, Zhu J, Zhou XG, Yuan WK (2007) Electrochem Commun 9:895
Rabinovich L, Lev O, Tsirlina GA (1999) J Electroanal Chem 466:45
Adekola FA, Diaw M, Colin C, Bauer D (1992) Electrochim Acta 37:2491
Sun L, Cao D, Wang G (2008) J Appl Electrochem 38:1415
Garbarino S, Burke S (2010) Int J Electrochem Sci 5:828
Chen M, Wang ZB, Zhou K, Chu YY (2010) Fuel Cells 10:1171
Moreira J, del Angel P, Ocampo AL, Sebastián PJ, Montoya JA, Castellanos RH (2004) Int J Hydrogen Energy 29:915
Toebes ML, van Dillen JA, de Jong KP (2001) J Mol Catal A Chem 173:75
Radkevich VZ, Senko TL, Wilson K, Grishenko LK, Zaderko AN, Diyuk VY (2008) Appl Catal A 335:241
Raróg W, Lenarcik I, Kowalczyk Z, Sentek J, Krukowski M, Zieliński J (2000) J Pol Chem 74:1473
Kowalczyk Z, Jodzis S, Raróg W, Zieliński J, Pielaszek J, Presz A (1999) Appl Catal A 184:95
Jackson SD, Shaw LA (1996) Appl Catal A 134:91
Pakuła M, Świątkowski A, Biniak S (1995) J Appl Electrochem 25:1038
Biniak S, Świątkowski A, Pakuła M (2001) In: Radovic LR (ed) Chemistry and physics of carbon, vol 27. Marcel Dekker Inc, New York, p 125
Świątkowski A, Pakuła M, Biniak S (1997) Electrochim Acta 42:1441
Biniak S, Pakuła M, Świątkowski A, Bystrzejewski M, Błażewicz S (2010) J Mater Res 25:1617
Alvares GF, Mamlouk M, Senthil Kumar SM, Scott K (2011) J Appl Electrochem 41:925
Senthil Kumar SM, Herrero JS, Irusta S, Scott K (2010) J Electroanal Chem 647:211
Czerwiński A, Kiersztyn I, Grdeń M, Czapla J (1999) J Electroanal Chem 471:190
Bucur RV, Bota F (1982) Electrochim Acta 27:521
Conway BE, Jerkiewicz G (1993) J Electroanal Chem 357:47
Liu JL, Lagger G, Tacchini P, Girault HH (2008) J Electroanal Chem 619–620:131
Biniak S, Diduszko R, Gac W, Pakuła M, Świątkowski A (2010) Reac Kinet Mech Cat 101:331
Barrera A, Viniegra M, Bosch P, Lara VH, Fuentes S (2001) Appl Catal B 34:97
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hofman, M., Świątkowski, A., Pakuła, M. et al. Cyclovoltammetric studies of carbon materials-supported palladium. Reac Kinet Mech Cat 106, 203–216 (2012). https://doi.org/10.1007/s11144-012-0425-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-012-0425-5