
Abstract
A GC/EIMS/SIM methodology has been developed to re-examine the path of carbon in photosynthesis. Exposing isolated spinach chloroplasts to 13CO2 on a solid support for a defined period followed by quenching and work-up provided a mixture of labelled sugar phosphates. After enzymatic dephosphorylation and derivatization, the Mox-TMS sugars were analysed using the above method. The purpose of the study was to try to calculate the atom% enrichment of 13C in as many of the individual carbons in each of the derivatized sugars as was practical using diagnostic fragment ions. In the event, only one 45 s experiment provided sufficient data to enable a range of enrichment values to be calculated. This confirmed that D-glycero-D-altro-octulose phosphate was present in the chloroplasts and was heavily labelled in the C4, C5 and C6 positions, in keeping with the hypothesis that it had an inclusive role and a labelling pattern consistent with a new modified pathway of carbon in photosynthesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In two previous papers, we described a GC/MS methodology, which is capable of providing a rapid, specific and quantitative means of analysis of 13C incorporation from 13CO2 into those sugar phosphates involved in the path of carbon in photosynthesis (Irvine et al. 1992; MacLeod et al. 2001). This provided a framework for a re-investigation of the photosynthetic carbon reduction (PCR) pathway in plants, commonly known as the Calvin Cycle (Calvin 1956). Although the PCR pathway as presented by Calvin (Fig. 1) is widely accepted, its re-examination is warranted for the following reasons: (i) the evidence upon which Calvin based the pathway was incomplete, (ii) more recent investigations support the inclusion of other sugar phosphates, notably octulose phosphates, in the pathway (Williams and MacLeod preceding paper), and (iii) the substantial advances in analytical technologies since Calvin’s data was collected in the 1950s, using 14C-labelling and paper chromatography, have the potential to provide more definitive and comprehensive results than were possible for Calvin to obtain.

The path of carbon in photosynthesis (Calvin 1956). Each asterisk (*) represents a carbon atom labelled during one turn of the cycle. For explanation of abbreviations, see preceding paper
This paper describes the measurement, using GC/EIMS/SIM, of 13C incorporation into individual carbon atoms of C4 to C8 sugar phosphates following short-term exposure of isolated chloroplasts to 13CO2 during PS.
Materials and methods
Chemicals
Sorbitol was purchased from Koch-Light, UK, sodium pyrophosphate from Mallinckrodt, USA and Percoll from Pharmacia, Sweden. Enzymes and cofactors were obtained from either Sigma, USA or Boehringer-Mannheim, Germany. Ion exchange resins were obtained from BioRad, USA. Analytical grade chemicals were obtained from Ajax, Australia, BDH, UK or Merck, Germany. All solvents were distilled before use while pyridine was pre-dried and distilled from calcium hydride under argon. Water was obtained from a Millipore Milli-Q system. The 13CO2 gas (99.9% atom enriched) was purchased from Cambridge Isotopes, UK and prepared to 1% in nitrogen in a gas cylinder. A second cylinder was made up with 1% unenriched CO2 in nitrogen. Gas analysis of the cylinders was carried out on a Varian 6000 gas chromatograph using a 1.83 m × 0.32 cm glass column packed with Porapak N 100–200 mesh stationary phase.
Plant material
Spinach (Spinacia oleracea, Yates Hybrid 102) seeds were obtained from Henderson Seed Co., Pty. Ltd., Lower Templestowe, Vic., Australia.
Growth of plant material; chloroplast isolation; chlorophyll assay; measurement of chloroplast intactness; polarographic measurement of chloroplast activity in suspension
The above methods have been described in detail in the accompanying paper (Williams and MacLeod preceding paper).
Measurement of chloroplast activity on filter membranes
The activity of the isolated chloroplasts mounted on a filter membrane was measured polarographically using a Clark oxygen electrode adapted for measurement of the oxygen evolution from leaf-discs (Delieu and Walker 1981). Methods for the preparation of chloroplasts for use in the leaf-disc oxygen electrode were based on those previously described (Cerovic et al. 1987). Chloroplasts were mounted on a filter membrane by filtering a suspension of chloroplasts through a cellulose nitrate membrane (Sartorius, 50 mm diameter, pore size 8 μm). This was accomplished using a Millipore solvent clarification apparatus fitted with a 100 ml funnel. A suspension of chloroplasts equivalent to 250 μg chlorophyll in 40 ml resuspension medium at pH 7.6 containing 330 mM sorbitol, 50 mM HEPES-KOH, 2 mM EDTA, 1 mM MgCl2 and 1 mM MnCl2 was allowed to filter through the membrane under gravity or with a very light suction so that the chloroplasts became trapped on the surface of the membrane in a thin film of resuspension medium. The chloroplasts were washed with a further 10 ml of the resuspension medium and then given a final wash with 3 ml of assay medium containing 6 mM inorganic phosphate, 22.5 U ml−1 alkaline phosphatase (Sigma, human placenta) and 1,000 U ml−1 catalase in resuspension medium. A disc of 1,018 mm2 (representing 94% of the chloroplasts) was cut from the membrane and mounted in a leaf-disc oxygen electrode (Hansatech). Once assembled, the leaf-disc oxygen electrode was charged with an atmosphere of 1% CO2 in N2 at atmospheric pressure and sealed. The chloroplasts were exposed to incandescent light from a pair of bifurcated fibre optic light sources at an intensity of 1,200 μEm−2 s−1 measured at the underside of the leaf-disc electrode light window. The temperature of the water bath supplying the water jacket, which regulated the oxygen electrode temperature was maintained at 23°C. After a lag period of approximately 1.5 min, the evolution of O2 could be monitored as a digital voltage on the oxygen electrode control box and was also recorded on a Goerz Metrawatt SE 120 chart recorder. The rate of O2 evolution reached a linear and maximum rate at approximately 4 min after switching on the lights. The rate was calculated from the linear portion of the trace.
Experiments in the leaf-disc oxygen electrode
Chloroplasts were prepared for experiments in the leaf disc oxygen electrode (as discussed for the assay of chloroplast activity on filter membranes in that equipment) and a linear rate of O2 evolution was established. At this time, a volume (60 ml) of 1% 13CO2 in N2 was flushed through the 6 ml chamber of the oxygen electrode that contained the membrane-mounted chloroplasts and after resealing the chamber, the chloroplasts were allowed to photosynthesize in the 13CO2-enriched atmosphere for a prescribed period of time before the reactions were terminated using the quenching methods described below. Experiments were conducted for 0, 30 and 45 s exposure to 1% 13CO2 in N2 and the “control” experiment was conducted with chloroplasts exposed to 1% 13CO2 in N2 for 45 s in the dark. In addition, a “blank” experiment was conducted in which the full extraction procedure was followed in the absence of chloroplasts to measure the extent of any contaminants that may interfere with the analysis. Five repetitions of each experiment were carried out using the same chloroplast preparation.
Reactions were terminated by dissembling the apparatus as quickly as possible and plunging the chloroplast-containing membrane into a beaker of liquid nitrogen. It was estimated that this procedure took no more than 2.5 s. The beaker containing the membrane was then stored in a freezer at −20°C where the liquid nitrogen was allowed to evaporate.
After overnight storage at −20°C the enzymes were denatured and the metabolites were extracted by removing the beaker containing the membranes from the freezer and immediately adding boiling 80% ethanol (75 ml) and boiling for a further 5 min. The membranes were then removed from the extract and thoroughly rinsed with water, adding the washings to the extract. After cooling, the pooled extracts were then evaporated to dryness at 37°C under a stream of dry nitrogen.
Design of a specific photosynthesis apparatus
Because of the limitations of the oxygen electrode for the type of experiments that were required, it was decided to design and construct an apparatus better suited for the purpose.
The basic requirements in the design and construction of the photosynthetic apparatus were (1) that the chloroplasts be mounted on a filter membrane inside a small chamber with a transparent window for the admission of light, (2) the filter membrane should be large enough to hold sufficient chloroplasts to provide enough extracted material for GC/MS analysis, (3) the atmosphere above the membrane must allow the establishment of steady state photosynthesis with unenriched CO2 and the rapid replacement of this with the same concentration of 13CO2 whilst causing minimum disturbance to the steady state, (4) the volume of the chamber above and below the membrane should be small in order to minimize the time taken to flush out the 12CO2 with 13CO2 using a reasonable gas flow rate. Also the volume of the chamber below the membrane must be small in order to minimize the “dead volume” of residual 12CO2, and (5) there must be provision for an appliance to inject a liquid to quench the photosynthetic reactions after a given time and for the convenient removal of the chloroplast extract after the quench.
Filter holders were available (Sartorius SM 165 08B) which, with minimal modification, provided a suitable basis on which to construct a prototype photosynthesis apparatus. The rapid replacement of unenriched CO2 with 13CO2 was provided for by the inclusion of two gas inlet ports each supplying an internal gas manifold around the outer perimeter of the chloroplast-supporting membrane. The manifolds directed 13CO2 across the entire membrane and drove excess gas out through a series of 6 gas outlet holes drilled in the housing above the centre of the membrane, which vented the expelled gases to the atmosphere. An electro-mechanical timer unit was constructed to permit the automatic and rapid switching of the gas supply from a gas cylinder containing 1% unenriched CO2 in nitrogen to another cylinder containing 1% 13CO2 in nitrogen via a pair of electrically operated gas valves, causing minimal disruption to the steady state of photosynthesis.
The inlet and outlet ports for the filter holder provided suitable ports for the entry and withdrawal of the quenching solution. A 10 ml syringe containing the quenching solution was attached to the filter inlet port via a tap, which could be opened to allow the injection of the quenching solution at the completion of the photosynthesis experiment. By applying a gentle suction via a tap at the filter outlet port, the extract could then be withdrawn into a flask containing boiling 80% ethanol located below the apparatus. The chloroplasts and apparatus were protected from the heat produced by the hotplate by the provision of a reflective heat shield. Light was provided via fibre optic cables, which greatly reduced the transmission of heat from the light source and reduced the need for cooling of the apparatus. The concentrations reported for chloroplast metabolites are variable but have usually been found to be in the range from 5 to 50 nmol per mg chlorophyll (Gerhardt et al. 1987; Giersch et al. 1980; Giersch 1979). A filter membrane loaded with chloroplasts equivalent to 250 μg of chlorophyll should therefore contain 1.25–12.5 nmol of most metabolites of interest. Assuming minimal losses during processing for GC/MS analysis, this was considered to be adequate chloroplast material for each experiment.
Experiments in the photosynthesis apparatus
Chloroplasts were prepared on a filter membrane as described for the experiments in the oxygen electrode. The loaded membrane was mounted intact in the photosynthesis apparatus. A gas mixture consisting of 1% unenriched CO2 in nitrogen was applied to the two gas inlet ports via an electrically operated valve connected to a specially made electro-mechanical timer unit. The gas stream (at a flow rate of 200 cm3 min−1) was humidified by bubbling it through a tube of water before entering the valve. The chloroplasts were illuminated through the transparent window in the apparatus by a pair of 150 W tungsten lamps, each directed through a bifurcated fibre optic light guide. The gas mixture was supplied for 4 min before the timer operated the valves supplying the gas and switched to the source supplying 1% 13CO2 in N2 for the prescribed period. Experiments were conducted for 0, 6, and 30 s exposure to 1% 13CO2 in N2 and the “control” experiment was conducted with chloroplasts exposed to 1% 13CO2 in N2 in the dark for 30 s.
The reactions were terminated and the metabolites were extracted by injecting hot 80% ethanol (5 ml) into the apparatus and allowing the chloroplasts to stand in the solution for 10 s. The ethanolic chloroplast extract was then drawn through the membrane using gentle suction into a flask that contained boiling 80% ethanol (30 ml). The extraction process was repeated with a further volume of boiling 20% ethanol (5 ml) followed by boiling water (5 ml). The combined extract was then boiled for 5 min and allowed to cool. After cooling, the extract was evaporated to dryness at 37°C using a stream of dry N2 gas.
Preparation of samples for GC/MS analysis
The dried samples from the photosynthesis experiments described above were redissolved in water (2 ml) and then processed for GC/MS analysis according to the following methods.
The chloroplast extract to be analyzed by GC/MS was applied to an Elut Bond strong anion exchange (SAX) solid phase extraction column (500 mg, Varian Associates, USA) and washed through with deionized water (30 ml). The eluate was discarded and the fraction containing both mono- and bisphosphates of the sugars was eluted with 0.5 M NH4HCO3 (5 ml) and collected in a polypropylene centrifuge tube (50 ml). Most of the buffer was removed by blowing the sample dry with a stream of dry nitrogen gas, whilst warming the sample in a water bath at 37°C. The samples were then dephosphorylated using human prostatic acid phosphatase, which was chosen for its ability to catalyze dephosphorylation quantitatively without causing the transformations produced by other phosphatases (Irvine et al. 1992). Dephosphorylation of up to 0.5 μmoles of sugar phosphates was carried out by the addition of 0.5 units of prostatic acid phosphatase to the sugar phosphate solution in 0.1 M ammonium acetate–acetic acid buffer pH 4.6 (typically 200 μl) and incubated for 24 h at 30°C. The reaction was terminated by heating at 100°C for 2 min.
Following dephosphorylation, the solutions containing the free sugars were deionized by stirring for 30 min with 0.5 g of mixed bed resin prepared using equal weights of anion (Bio-Rad AG 1 × 8, 200–400 mesh, HCO −3 form) and cation (Bio-Rad AG 50 W × 8, 200–400 mesh, H+ form) exchange resins together with deionized water. The resin was removed by filtration through a small column fashioned from a pipette tip (5 ml) plugged with cotton wool and containing a further 0.25 g of the mixed bed resin. The sample was washed through with water (15 ml), collected in a polypropylene centrifuge tube (50 ml) and lyophilized. The resulting residue was washed with deionized water (1 ml) into an Eppendorf tube (1.5 ml) and again lyophilized. The residue was redissolved in water (120 μl) and the entire sample was transferred to a Reacti-Vial (200 μl) and dried over P2O5. The sugars were subsequently dried under high vacuum for 1 h.
Derivatization of dephosphorylated sugar phosphates
Sugars were prepared for GC/MS analysis as the methoxime-TMS derivatives using methods similar to those first described by Sweeley (Sweeley et al. 1963) for 1 mg scale derivatizations. In our investigation, smaller-scale derivatizations were required. Up to 350 nmol (approx. 50 μg) of the dried sugars in Reacti-Vials (0.2 ml) were derivatized by the addition, under dry nitrogen, of 1% methoxylamine in pyridine (40 μl) and refluxing at 80°C for 2 h in a Reacti-therm heater. The mixture was cooled to room temperature then silylation was accomplished by the addition under dry nitrogen, of 10 μl of Regisil (Pierce) followed by heating for 0.5 h at 80°C. The mixture was cooled to room temperature then diluted as appropriate with dry pyridine for GC/MS analysis. Injections of 1 μl ideally contained no more than 125 pmol of the sugar under analysis in order to avoid saturation of the mass selective detector, as determined by sensitivity runs with derivatized glucose.
GC/MS analysis of derivatized sugars
Derivatized sugars were analyzed by capillary GC/MS on a Hewlett Packard 5890 gas chromatograph interfaced with a Hewlett Packard HP5970B MSD and controlled by a Hewlett Packard HP59970C ChemStation. Gas chromatography was performed on an HP-1 capillary column (12.5 m × 0.2 mm i.d., 0.33 μm cross-linked methyl silicone stationary phase) in the splitless mode with helium carrier gas (flow rate 1 ml min−1). The column temperature was held at 100°C for 2 min with the filament off for the passage of the solvent and excess reagent through the column. The temperature was then increased to 250°C at 10°C min−1, and held at 250°C for 3 min. Spectra were obtained by electron impact (EI) ionization at 70 eV. Full scan spectra of the derivatized sugars were usually obtained by scanning from m/z 100 to 660 at a rate of 0.77 scans s−1 and represent an average of scans taken across the top of each relevant chromatographic peak. Selected ion monitoring (SIM) experiments on clusters of ions of interest in the spectra of the derivatized sugars were carried out in groups of 8–12 ions per peak with dwell times of 30 ms per ion. All results represent the mean of at least three separate injections unless otherwise stated. Corrections for natural abundance isotopes of C, H, N, O and Si and contributions from adjacent ions one mass unit lower of comparable intensity were made in calculating 13C enrichments in individual ions. Only fragment ions which had been found to have essentially a single origin as the result of the GC/MS analysis of specifically 13C-labelled sugar standards were used in this study (Irvine et al. 1992; MacLeod et al. 2001).
Identification of sugar phosphates in chloroplast extracts
The individual unlabelled sugar phosphates present in the chloroplast extracts were identified by the retention times and full scan mass spectra of their dephosphorylated Mox-TMS derivatives on the above GC/MS system when compared with those of authentic sugars run under identical conditions (Irvine et al. 1992; MacLeod et al. 2001).
Quantification by GC/MS of sugar phosphates in chloroplast extracts
Quantification of analytes is best achieved by the inclusion in the sample mixture of a known amount of an internal standard having similar chemical and physical characteristics to those of the analytes.
Ideally, for GC/MS analysis the internal standard should be a stable isotope analogue of the analyte under investigation and when there are multiple analytes, a stable isotope of each analyte is preferred. In this investigation however, the analytes (derivatized sugars) are themselves expected to become multiply-labelled with the 13C isotope during photosynthesis and the stable isotope dilution method therefore could not be used. The alternative approach was used of adding a compound that is similar to the sugars under investigation but is unlikely to be encountered in chloroplasts and would be well separated from the analytes on GC/MS.
For SIM, the mass spectrum of the derivatized standard chosen for this GC/MS investigation needed to contain an ion which was common to all of the sugars being measured. In addition, this ion must not become labelled in a 13CO2 experiment. The sugar alcohols seemed to be an obvious choice. The mass spectra of the per-TMS derivative of these compounds show a prominent m/z 147 ion representing the pentamethyldisiloxonium ion ([(CH3)3SiOSi(CH3)2]+), as do the mass spectra of all of the sugar methoxime TMS derivatives. Since this ion does not contain any carbons derived from the sugar, it cannot become labelled during a 13C isotope incorporation experiment. Thus, SIM of the m/z 147 ion was the method of choice for quantitation studies.
Erythritol was chosen as the internal standard because (1) the m/z 147 ion of its per-TMS derivative gave a linear and reproducible response when calibrated against the same ion in the mass spectra of Mox-TMS sugars at levels up to 120 pmol and (2) it was well separated on the HP-1 GC column from the sugars under consideration.
Sugars which were available in the free form with a high degree of purity were dried for 3 days over phosphorous pentoxide under a low vacuum. Approximately 0.02 g of the dried powder or syrup (ketopentoses) was weighed to five decimal places to prepare standard solutions of each sugar. Appropriate dilutions of these were taken to prepare a set of 0.5 mM standard solutions.
Some sugars were only available as the phosphate esters and frequently in the presence of various amounts of buffer salts. In these cases, after weighing the dried powders and preparing standard solutions, they were assayed enzymically to establish their concentrations prior to use. Sedoheptulose, D-glycero D-ido- and D-glycero D-altro-octuloses were assayed as their bisphosphates using an aldolase-based assay (Bergmeyer and Bernt 1974). Erythrose was assayed as the monophosphate ester using a transketolase-based assay (Paoletti et al. 1979). Sugar phosphates (3 μmol) were then evaporated to dryness in a rotary film evaporator and dephosphorylated in a solution (0.5 ml) which contained citrate buffer 50 μmol, pH 5.4) and human prostatic acid phosphatase (1.25 units). After allowing the dephosphorylation to proceed for 4 h at 37°C, the reaction was terminated by heating at 100°C for 2 min. The samples were then treated with mixed bed resin and lyophilized.
Recovery from the dephosphorylation step was assessed by carrying out parallel dephosphorylations of glucose 6-phosphate, fructose 6-phosphate and fructose 1,6-bisphosphate, all of which were assayed enzymically as the free sugars. The recovery of the monophosphates was 88% and of the bisphosphate was 78%. These figures provided a useful estimate of the likely recoveries of those sugars (above), which could not be assayed in the non-phosphorylated form.
Appropriate dilutions of the lyophilized sugars were then prepared to produce a set of 0.5 mM solutions for use as standards. Groups of sugar mixtures were prepared, each containing 500 μl of the 0.5 mM standard solution. Each sugar was thus present in a volume of 2 ml and at a concentration of 0.125 mM. The first group contained erythrose, fructose and D-glycero D-ido-octulose. The second group contained glucose, sedoheptulose and D-glycero-D-altro-octulose. Pentoses were calibrated separately. A series of dilutions of the standard mixtures containing 0.025, 0.125, 1, 2, 3, 4 or 5 nmol of each sugar together with 5 nmol of the erythritol standard were added to separate ReactiVials.
The mixtures were then first dried over P2O5 then under high vacuum and derivatized (Mox-TMS). This gave solutions which contained 0.5, 2.5, 20, 40, 60, 80, or 100 pmol of each sugar together with 100 pmol of the erythritol standard in each μl of the derivatized solution.
GC/MS analysis was carried out by the measurement of the peak areas obtained for the m/z 147 ion of each sugar using single ion monitoring. Three injections were made at each sugar level and the average ratio of sugar to erythritol peak area was determined. In those cases where the Mox-TMS derivative of the sugar showed peaks for both syn- and anti-forms, the area of the leading peak was used. A graph plotting the measured ratio of each sugar m/z 147 ion area to the erythritol m/z 147 ion area against the actual ratio of the sugar to erythritol for the series of sugar concentrations was then plotted. The slopes of these graphs then represent the calibration factors relating the sugars to the internal standard, erythritol, and were determined as the slope of the least squares line of regression of measured area ratio on actual ratio (Table 1).
Results and discussion
Initial experiments were carried out using the leaf-disc oxygen electrode during the time that the special photosynthesis apparatus was being constructed in-house. Obvious disadvantages inherent in this approach were (1) the difficulty in achieving complete, instantaneous displacement of 12CO2 with 13CO2 and (2) the time taken to dissemble the apparatus and quench the photosynthesis reaction after exposure of the chloroplasts to 13CO2, leaving open the possibility of partial back exchange of the 13C-labelled sugars. One advantage was that the photosynthetic activity of the chloroplast preparations could be measured in situ prior to carrying out the 13CO2 exchange experiments.
There were several pitfalls that had to be overcome before PS exchange experiments with 13CO2 could be performed. Foremost of these was the low concentrations of sugars of the PS pathway observed in the chloroplast extracts in initial experiments using unlabelled CO2, together with the presence of other sugar-like components in the GC/MS analysis . The latter was eventually traced to adsorption and decomposition of the small quantities (pmol) of sugar phosphates present in the chloroplasts on the surfaces of glass vessels used during the workup procedure. To minimise decomposition, these were replaced where possible with non-glass vessels. Losses due to adsorption could not be completely avoided and necessitated combining extracts from five consecutive experiments (using the same chloroplast preparation) in order to try to obtain useful data on those sugars present in lowest concentrations. Contaminants introduced during the workup presented another problem in the GC/MS analysis, which required extensive investigation of their sources. The only contaminant that could not be completely eliminated was sorbitol but this did not interfere with the sugar analysis as the retention time of its TMS derivative differed from those of the Mox-TMS sugars under investigation.
The experiment carried out in the oxygen electrode at zero time exposure of the chloroplast preparation to 1% 13CO2 in nitrogen showed, after work-up, no incorporation of label in the sugars. As well, the “control” experiment carried out with chloroplasts exposed to 1% 13CO2 in the dark for 45 s produced no 13C enrichment in any of the sugars in the chloroplast extract.
After analysis of the results of four experiments carried out in the oxygen electrode, involving exposure of the chloroplasts to 13CO2 for 30 s (twice) and for 45 s (twice), only one of the 45 s experiments produced a reasonably comprehensive set of results (Table 2). The main factor contributing to the inadequacy of the remaining experiments was the lack of sufficient material to enable GC/MS/SIM runs to be performed on all of relevant ion clusters as set out in a previous paper (see Table 2, MacLeod et al. 2001).
Experiments were then carried out using the custom-made photosynthesis apparatus, involving exposure of isolated chloroplasts to 1% 13CO2 in N2 for 30 s in the dark and for 0, 6 and 30 s under controlled light conditions. After work-up, both the “dark” experiment and zero time exposure showed no 13C incorporation in any of the sugars analysed. As was the case for all but one of the oxygen electrode experiments, the 6 s and 30 s experiments after work-up did not contain a sufficient concentration of the sugar Mox-TMS derivatives, even after combining five experiments, to enable selective ion profiles to be obtained on all of the relevant ions in all of the sugars. This was particularly disappointing as the specially designed apparatus performed well otherwise.
In all of the above experiments, the concentrations of the sugar phosphates present in the chloroplast extracts were determined by SIM of the m/z 147 ion present in the mass spectra of the dephosphorylated Mox-TMS derivative, using erythritol as internal standard (see Materials and methods). A correction was made for losses incurred during the chloroplast work-up and dephosphorylation stages by adding 14C-labelled glucose 6-phosphate to a single membrane-mounted chloroplast preparation immediately prior to quenching in the special apparatus. The measured recovery figure of 73% of 14C-labeled glucose represents a minimum recovery value since the method eventually used involved the pooling of the extracts from five membranes, which would be expected to lead to reduced losses. Table 3 lists the values obtained. As expected, the two “blank” runs showed only traces of glucose, fructose and pentoses while the 45 s “dark” experiment showed lower than normal levels of all sugars except glucose. The other six experiments, five using the oxygen electrode and one using the special apparatus, showed sugar concentrations which, although varying from experiment to experiment, were of the same order of magnitude to those previously reported (Lilley et al. 1977).
No C3 sugars could be analysed under the GC conditions used since they eluted with the residual derivatizing agents. A tetrose sugar was present which had the same mass spectrum and elution time as erythrose (Table 3) but subsequent analysis of label incorporation into selected ions showed no 13C enrichment. The unidentified heptulose listed in Table 3 did show very significant 13C incorporation in the few ion clusters measured (Table 2), comparable to that observed in sedoheptulose. D-g-D-a-Octulose was identified by the retention times of its syn- and anti-isomers but the earlier eluting D-g-D-i-octulose was not detectable.
The enrichment results obtained in Table 2 were sufficient to calculate atom% excess in some but not all of the individual carbon atoms of the sugars listed (see Table 2 in MacLeod et al. 2001). For example, the enrichment in C-2 of ribose (23.8%) was obtained by subtraction of m/z 307 (C3-5) from m/z 319 (C2-5). Similarly, subtracting the 13C enrichments in the ions m/z 307 and 205 (C4-5) gave the atom% excess in C3. C-1 (12.1%) was calculated by subtracting the above value for C2 from the enrichment in the m/z 160 ion (C1-2). In some cases where it was not possible to obtain enrichment data for individual carbon atoms, values for consecutive two carbon units were calculated.
While it is obvious that the results obtained fall short of the objective, for reasons given above, they nevertheless allow some pertinent observations to be made. The presence of D-g-D-a-octulose in isolated chloroplasts at concentrations between 0.2 and 3.5 nmol (mg chl)−1 is unequivocally confirmed (Table 3). In the 45 s experiment, the atom% 13C excess in carbons 4, 5 and 6 of the octulose is between 25% and 30%, comparable with the values calculated for C-2 and C-3 of ribose, C-3 and C-4 of fructose and glucose, and C-4 and C-5 of sedoheptulose (Table 4). These sugars and their above carbons are implicated in CO2 fixation in Calvin’s photosynthetic pathway (Fig. 1) and it is therefore not unreasonable to speculate that D-g-D-a-octulose also has a role in this pathway, in support of the conclusions drawn in the preceding paper (Williams and MacLeod 2006). The pattern of 13C labelling of carbons 4, 5 and 6 of D-g-D-a-octulose presented here is precisely the same as that shown in the octulose 1,8-bisphosphate by NMR spectroscopy of a spinach leaf extract after photosynthesis in 13CO2 (Bartlett et al. 1989).
A further interesting observation was the presence of a second heptulose sugar, albeit in much lower concentrations than that of sedoheptulose (Table 3). It had a substantial atom% excess at C-4 of 54.2%, comparable with that observed for the same carbon in sedoheptulose. A GC/MS comparison of the retention time of its Mox-TMS derivative with that of mannoheptulose eliminated this as a possibility and so it remains unidentified.
One of the anomalies of the Calvin Pathway (Fig. 1) has been the failure to identify erythrose phosphate in chloroplasts, even though it is proposed to play an integral role in CO2 fixation. In this study, although we were able to detect a four carbon sugar with the same retention time and mass spectrum as Mox-TMS-erythrose (Table 3), GC/MS/SIM analysis showed no incorporation of 13C in any of its fragment ions. It could therefore be assumed that the level of erythrose phosphate participating in the pathway is below that detectable by GC/MS/SIM. However, the identification and evidence for the presence of Ery 4-P in the Calvin and Pentose pathways has always been weak and contentious (Williams et al. 1980).
Unfortunately, circumstances have not allowed us to repeat the above experiments on a scale which could provide sufficient amounts of sugar phosphates to allow more complete data on the time-course of 13C incorporation from 13CO2 during photosynthesis to be obtained. In support of an earlier study of gluconeogenesis in liver cells (Desage et al. 1989) it has, however, been shown here that GC/MS/SIM offers a viable alternative methodology for the study of positional isotope analysis of intermediates formed from stable-isotope labelled precursors in complex biological systems.
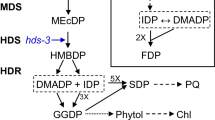
In summary, the results reported here and in the preceding paper have established the formation and specific predictive labelling of octulose phosphate(s) in spinach chloroplasts by 13CO2 and 14CO2 during photosynthesis. The data do not unequivocally prove that octulose phosphates are direct and exclusive intermediates of a new set of partial reactions for the path of carbon that were inadvertently missed by Calvin and his colleagues. However we suggest that octulose phosphate formation may fulfil a role as mono- and bisphosphates in a shunt (as defined by Newsholme and Leech 1983) that is attached to the Calvin reaction scheme (Fig. 1). Figure 2 is a structural display of the new reaction scheme presented in the accompanying paper. The shunt shows that the octulose phosphate reactions intersect with and may in time and space compartmentalize Seh 1,7-P2 from SBPase and thereby limit its important role in the regulation of flux in the carbon path of PS.

An extension of the Calvin pathway by the inclusion of an octulose phosphate shunt (vide infra). Each asterisk (*) represents a carbon atom labelled during one turn of the cycle. Sugar phosphates in red represent those participating in the octulose shunt. For explanation of abbreviations, see preceding paper
References
Bartlett MRE, Collins JG, Flanigan IL, MacLeod JK, Williams JF (1989) CO2 labelling of octulose bisphosphates during photosynthesis. An NMR study using intact spinach leaves. Biochem Int 18:35–46
Bergmeyer HU, Bernt E (1974) Fructose-1,6-diphosphate aldolase. UV assay, manual method. In: Bergmeyer HU (ed) Methods of enzymatic analysis, vol 2. Second English Edition, Academic Press, New York, pp 1100–1105
Calvin M (1956) The photosynthetic carbon cycle. J Chem Soc (London):1895–1915
Cerovic ZG, Cheesbrough JK, Walker DA (1987) Photosynthesis by intact isolated chloroplasts on solid support. Plant Physiol 84:1249–1251
Delieu TJ, Walker DA (1981) Polarographic measurement of photosynthetic O2 evolution by leaf discs. New Phytol 89:165–175
Desage M, Guilly R, Brazier J-L, Riou J-P, Beylot M, Normand S, Vidal H (1989) Positional isotopic analysis of labelled glucose by mass spectrometry: applications to the study of gluconeogenesis in liver cells. Biomed Environ Mass Spectrom 18:1010–1015
Gerhardt R, Stitt M, Heldt HW (1987) Subcellular metabolite levels in spinach leaves. Regulation of sucrose synthesis during diurnal alterations in photosynthetic partitioning. Plant Physiol 83:399–407
Giersch C (1979) Quantitative high-performance liquid chromatographic analysis of 14C-labelled photosynthetic intermediates in isolated intact chloroplasts. J Chromatogr 172:153–161
Giersch C, Heber U, Kaiser G, Walker DA, Robinson SP (1980) Intracellular metabolite gradients and flow of carbon during photosynthesis of leaf protoplasts. Arch Biochem Biophys 205:246–259
Irvine RW, Flanigan IL, MacLeod JK, Collins JG, Williams JF (1992) Mass spectrometric studies of the path of carbon in photosynthesis: positional isotopic analysis of 13C-labelled 2-octulose phosphates. Org Mass Spectrom 27:1052–1060
Lilley RM, Chon CJ, Mosbach A, Heldt HW (1977) The distribution of metabolites between spinach chloroplasts and medium during photosynthesis in vitro. Biochem Biophys Acta 460:259–272
MacLeod JK, Flanigan IL, Williams JF, Collins JG (2001) Mass spectrometric studies of the path of carbon in photosynthesis: positional isotopic analysis of 13C-labelled C4 to C7 sugar phosphates. J Mass Spectrom 36:500–508
Newsholme EA, Leech AR (1983) Biochemistry for the medical sciences. John Wiley & Sons, New York, p 334
Paoletti F, Williams JF, Horecker BL (1979) An enzymic method for the analysis of D-erythrose 4-phosphate. Anal Biochem 95:250–253
Sweeley CC, Bentley R, Makita M, Wells WW (1963) Gas-liquid chromatography of trimethylsilyl derivatives of sugars and related substances. J Am Chem Soc 85:2497–2507
Williams JF, Blackmore PF, Duke CC, MacLeod JK (1980) Fact, uncertainty and speculation concerning the biochemistry of D-erythrose 4-phosphate and its metabolic roles. Int J Biochem 12:339–344
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Flanigan, I.L., MacLeod, J.K. & Williams, J.F. A re-investigation of the path of carbon in photosynthesis utilizing GC/MS methodology. Unequivocal verification of the participation of octulose phosphates in the pathway. Photosynth Res 90, 149–159 (2006). https://doi.org/10.1007/s11120-006-9114-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-006-9114-4