
Abstract
Purpose
Neglected tropical diseases (NTDs) represent are a heterogeneous group of communicable diseases that are found within the poorest populations of the world. There are 23 NTDs that have been prioritized by the World Health Organization, which are endemic in 149 countries and affect more than 1.4 billion people, costing these developing economies billions of dollars annually. The NTDs result from four different causative pathogens: protozoa, bacteria, helminth and virus. The majority of the diseases lack effective treatments. Therefore, new therapeutics for NTDs are desperately needed.
Methods
We describe various high throughput screening and computational approaches that have been performed in recent years. We have collated the molecules identified in these studies and calculated molecular properties.
Results
Numerous global repurposing efforts have yielded some promising compounds for various neglected tropical diseases. These compounds when analyzed as one would expect appear drug-like. Several large datasets are also now in the public domain and this enables machine learning models to be constructed that then facilitate the discovery of new molecules for these pathogens.
Conclusions
In the space of a few years many groups have either performed experimental or computational repurposing high throughput screens against neglected diseases. These have identified compounds which in many cases are already approved drugs. Such approaches perhaps offer a more efficient way to develop treatments which are generally not a focus for global pharmaceutical companies because of the economics or the lack of a viable market. Other diseases could perhaps benefit from these repurposing approaches.
Similar content being viewed by others

Introduction
Since the 1980’s the pharmaceutical industry has taken advantage of the systematic advances in molecular biology/genetic engineering and replaced phenotypic and whole-cell High Throughput Screening (HTS) with target-based screening assays (1). While target-based screens were developed that utilized simple recombinant protein enzyme assays provided many advantages in terms of cost and scalability, they relied on the assumption that the selected target is in fact the best and most druggable target for a given disease. However, in the last decade, we have seen a clear shift back towards using phenotypic screens as an initials starting point for drug discovery, especially for neglected diseases for which the drug targets are generally poorly understood or where target-based approaches failed (1,2). An analysis of the origin of first-in-class small molecules proposed that phenotypic screens identified more novel inhibitors than other approaches between the years 1999 and 2008 (3). However, phenotypic screening has the disadvantage that it does not provide information about the target/s for a hit.
Neglected Tropical Diseases (NTDs) are a diverse group of 23 predominantly communicable diseases (Table I) that prevail in tropical and subtropical conditions, affecting 149 countries, more than 1 billion people in the developing world (4,5), and costing those global economies billions of dollars every year. These 23 NTDs disproportionately impact poorer regions and contribute to substantial morbidity, disability and poverty in many low- and middle-income countries (6). It is important to realize that in many cases these pathogens are eukaryotic parasites which have complex life cycles and varied mechanisms for evading their host immune system. In addition, some of these parasites are not genetically tractable in the laboratory which further complicates drug discovery efforts. The inability to translate many of these pathogens into a lab setting is one reason for the lack of research into these diseases. Perhaps more critical is the absence of a lucrative market for NTD treatments. These diseases generally lack market incentives to spur product development (7,8,9). As a result, NTDs receive relatively little research investment ($80M - $500M / year) from governments or pharmaceutical companies, compared with the billions of dollars invested in diseases such as cancer and heart disease or other blockbuster diseases (10). Although these neglected diseases continue to lack major economic incentives due to their financially unattractive affected population, (7,8,9) there is a tropical disease priority review voucher awarded to companies that successfully develop FDA-approved treatments for particular diseases (11,12,13) (Table II) some of which overlap with the WHO definitions of tropical neglected diseases (Table I). This is anticipated to assist companies to focus on developing treatments for these diseases. Another way to overcome the challenges of NTD drug development is to reposition or repurpose existing drugs (15,16,17,18,19,20,21,22). Utilizing compounds that have already been approved by the FDA, compounds with known physicochemical and absorption, distribution, metabolism, excretion and toxicology (ADMET) properties, can accelerate the development process of an old drug through financial and regulatory hurdles to market. The efficiency of a repurposing approach is especially critical for NTDs which typically do not have viable commercial products already approved or treatment paths that are economically feasible. In addition to the potential monetary benefits, the development of a repositioned product can shorten the timeframe for drug discovery from 10-17 years to 3-12 years which is especially important in cases where the spread of disease is rapid and costly (23,24).
Despite the benefits of drug repurposing, pharmaceutical companies do not seem to be making this standard practice as yet. ‘Use patents’ are not as valuable as ‘synthesis patents’ and since repurposing is almost by definition exclusively limiting to ‘use patents’, it is more beneficial for pharmaceutical companies to invest their financial resources in their own research and development of new compounds, potentially overlooking effective drugs that already exist. For small drug companies and biotech firms, it is more urgent to build valuable IP in order to compete with larger companies and so they are even less likely to find value in repurposing drugs. Due to the poor economic tradeoffs, a lot of repurposing efforts are being championed by academia. However, there are many recent examples of successful collaborations between industry, government, and academia, where large libraries of compounds are made available for high throughput screening including by Novo Nordisk (25), Novartis (26), GlaxoSmith Kline (27), Pfizer (28), and other companies as parts of consortia (e.g. More Medicines for TB (29,30), TB Drug Accelerator, NIH NCATS etc).
In addition to industrial libraries, repurposing studies benefit from any compound libraries that are accessible. These include NCATS (31) /NIH (32,33), Microsource (34,35,36), LOPAC (37), Prestwick (38), Broad (39), Calibr (40,41), bioactive libraries (42), small molecule libraries (43), collections of natural products (42), and others. The library that arguably provides the best benefit is the FDA approved library (44,45,46,47,48). Because this list seems to be updated sporadically and obtained from different sources, studies that refer to the library of FDA approved drugs describe a library ranging from 774 (45), to 1,012 (48) to 1,766 compounds (49). Johansen et al. extended this library to screen a combined 2,600 US and “non-US” approved drugs in one study (50), and a combination of FDA approved drugs and “ex-US-approved drugs” in another (51). Although diseases occurring throughout the globe can be impacted by these types of repurposing efforts, there are no studies identified that have screened libraries compiled by regulatory bodies of other countries with the intent of repurposing drugs for a neglected disease to our knowledge. While these regulatory bodies exist, they seem to be overlooked by most researchers trying to repurpose drugs for NTD’s and the majority of “approved” compound screening uses the US FDA library.
Data Mining Approach
While tremendous progress has been made in neglected disease drug discovery over the last few years (52), there are still many gaps to be addressed (53). These diseases and variations of their associated terminology were searched in the literature using multiple search engines along with “high-throughput screen” and “identified” to find those papers in which screening was performed with some success. The ~7000 rare diseases often discussed alongside neglected diseases (54) were not considered for this study. The use of the Medicines for Malaria Ventures (MMV) box has been extensive and only a handful of those studies are presented here. Van Voorhis et al have a more complete compilation of that work (55). There are also considerable efforts in the development of assays (56,57,58,59,60,61), lead optimization (62,63), mechanism of action elucidation (64), and smaller scale repurposing studies for neglected diseases (65,66,67,68,69,70,71), but these were outside the scope of this investigation. Due to the number of people living with these neglected diseases and the urgency needed to respond to their emergence, studies involving high throughput screening for Dengue, Zika, and Ebola viruses were included as recent examples. In addition to presenting some of the work that has been done in the NTD repurposing field, we have compiled a database of over 1,200 compounds suggested for repurposing by the authors developed across the many studies summarized herein.
Chagas Disease
Chagas disease is a NTD that is known to be caused by the kinetoplastid parasite Trypanosoma cruzi (72). The disease is a problem in Latin America where it is endemic, but it is also increasingly found in North America and Europe, which it reaches through immigration (73,74,75). The clinical signs of Chagas diseases are manifested in different phases. In all cases, the disease ultimately leads to death and the only available contingency plans rely on organ transplantation. The spread of Chagas disease to North America and beyond is highlighting the need for novel, safe, and effective therapeutics to treat T. cruzi infection. Both target-based and phenotypic-based studies have been performed in recent years to address this.
De Rycker et al. employed a novel screening cascade to screen two libraries, the SelleckChem, which is comprised of 421 FDA-approved drugs and the NIH Clinical Collection, which consists of 727 compounds tested in clinical trials ((76), (77)). The drug concentrations were 5 and 15 μM. The cascade was made up of an initial single-point primary screen, that was followed by a high content screening assay to determine potency, a static-cidality and rate-of-kill assessment, as well as an in vitro CYP51 activity determination (to measure the inhibition of agonist-induced calcium flux). Inhibitors that were found to interfere with calcium homeostasis were shown to have moderately slow killing profiles despite their high potency. The remaining hits were classified as central nervous system (CNS) targeting lipophilic amines. Based on potency, selectivity and cidal profiles, the most interesting drugs were determined to be ifenprodil, ziprasidone, clemastine, clofibrate and azelastine. All compounds have reasonable bioavailability but are currently limited by toxicity. Future work would have to be performed to eliminate the CNS penetration or H1 antagonism to make the compounds more suitable for treatment of T. cruzi in human targets (77). During the development of the above mentioned CYP51 assay, Riley et al., identified several hits mentioned briefly in a separate paper (78). They used the CYP51 enzyme of the Chagas parasite expressed in E. coli as a way to distinguish compounds with similar mechanisms of actions from those used by current drugs where resistance is a growing problem. 200,000 compounds were initially screened for their activity and the resulting hit compounds were represented by 129 compound classes. Compounds were de-prioritized based on chemical similarity to known treatments and the remaining hits, represented by 5 singletons, were suggested for repurposing (78).
Keenan et al. utilized a luminescence cell-based HTS in order to identify inhibitors of T. cruzi replication (39). The selected compounds were screened based on their ability to inhibit intracellular trypanosomes from replicating inside the host cells. The surviving parasites were then assessed via detection of a luminescent reporter after cells were lysed. This group screened over 300,000 compounds, and over 4,000 were identified (hit rate ~1.4%). Compounds from this list were then prioritized based on the following characteristics: homology to known antifungal pharmacophores with anti-trypanosomal activity, compounds that contain a basic heterocycle and two additional lipophilic or aromatic moieties in a trigonal or tetrahedral arrangement around a central atom, synthetic tractability, novel structures to the field, and the extent to which structural domains influenced the activity. Fifteen lead hit structures were initially identified, and based on the mentioned criteria, seven were identified. In vitro activity studies against Trypanosoma cruzi and in vivo studies for toxicity further filtered the candidates down to CM100 (IC50 = 0.169 uM) and CM74 (IC50 = 0.075 μM) as promising therapeutics for Chagas disease (39).
In 2014 Planer et. al. suggested two combination therapies for the treatment of Chagas disease; amlodipine and clemastine, both in combination with posaconazole (36). Most drugs screened did not have dramatic effect alone, with the exceptions of benznidazole and posaconazole. However, it should be noted that many compounds were not ineffective in vitro because investigators intentionally used low dosages in order to observe the effects on bloodstream parasitemia (36).
From the genome of T. cruzi, several enzymatic pathways were identified as potential targets and compounds with associations to those pathways were investigated in a Bayesian model that considered 300,000 small molecules from previous studies. After in silico screening, 97 compounds were screened in vitro, 11 were found to be active and several were found to be quite effective in vivo in mice, with one compound, pyronaridine (85% efficacy) selected for further investigation (79).
To find new inhibitors of the cruzain enzyme present in Trypanosoma cruzi, Wiggers et al. screened the ZINC database for compounds with lead-like properties and then performed consensus scoring using both target-based molecular docking, and ligand based similarity searching (80). This narrowed 8.5 million compounds to 8,600 compounds, which were then screened through Glide XP and HQSAR. The top 5% of compounds that passed both screens were visually inspected and 23 compounds were selected for in vitro testing. The top two compounds from this screen were structurally characterized by X-ray crystallography to understand their binding modes with cruzain (80).
Gunatilleke et al. screened over 100,000 compounds from multiple libraries and observed that several compounds that contain similar structural features were effective against Chagas disease using CYP51 as a permissive target for the small molecule assay (43). Some 185 of the top hits were nitrogen-containing aromatic heterocyclic pharmacophores. Of these, 92 compounds had the R group in the para position and the nitrogen in meta position within a ring. These compounds have been added to the ZINC database for future use, but no in vitro verification has been performed yet (43).
Unfortunately, due to the complexity of the disease, the various stages in parasitic lifecycle, and the inherent differences between human patients and in vivo, in vitro, and in silico studies, compounds which show great promise in pre-clinical studies do not necessarily translate to effective clinical approaches (81). For Chagas disease this was evident in the case of posaconazole which failed to maintain sustained efficacy over the course of a 60 day clinical trial (82). In contrast, benznidazole (a currently utilized treatment), was shown in a separate study to have 82% sustained response after a year (83). Unfortunately, benznidazole has known side effects which can limit its broad use (81). A major project by the DNDi to find superior treatment options has been updated to filter out compounds with CYP51 inhibitors like posaconazole and to better understand rate of kill to predict slow acting versus fast acting compounds (81).
Human African Trypanosomiasis (HAT)
Human African Trypanosomiasis (HAT) is another kinetoplastid disease that is found in sub-Saharan Africa, where 70 million people are at risk of this disease and an estimated 30,000 are currently infected (84). HAT is known to be caused by two different subspecies of the protozoan parasite Trypanosoma brucei. While there are treatments for this disease HAT drugs have historically had limitations relating to their administration, toxicity or cost. There are relatively few alternative drugs or validated drug targets which is driving research to identify parasite-specific antitrypanosomal lead compounds. Hence there have been efforts to develop a fluorometric viability assay for HTS (85,86,87,88,89,90,91,92,93,94,95,96,97,98). In a successful example of a partnership between industry and academia, 42,444 compounds were provided by GlaxoSmithKline and screened by Diaz et al., for growth inhibition of Trypanosoma brucei (27). 797 compounds showed activity against this sleeping sickness parasite while also not presenting signs of toxicity to human cells (27). Further analysis was performed to distinguish cell death vs. cell growth inhibition and 129 compounds that cause parasite death were grouped into clusters based on structural similarity (27).
Blaazer et al., investigated a library of compound fragments that efficiently covered chemical space as defined by both complexity and number of rings (99). Beginning with 1,040 compounds, they screened the compounds in vitro against a phosphodiesterase (PDE) present in T. brucei and an ortholog from humans. The fragments displayed activity in each assay with only 7 fragments showing selectivity (99). The authors focused on 4 fragments with structural similarities to known drug scaffolds and 32 associated analogs in further phenotypic assays for parasites causing sleeping sickness, Chagas, leishmaniasis, and malaria, as well as human cells for selectivity consideration (99).
In 2011, Piclamilast was identified by Bland et al. as a phosphodiesterase (PDE) inhibitor potentially useful against HAT (100). Their approach involved an interesting method in which they aimed to repurpose knowledge about phosphodiesterase inhibitors from human-focused medicinal chemistry and to leverage this understanding into a treatment for the HAT parasite. They identified piclamilast (a known PDE inhibitor in humans) as a relevant starting place for optimization. This was followed by a comparison of piclamilast docking into the human and parasitic binding pockets as well as an SAR study of analogs.
Leishmaniasis
This parasite is transmitted by the sand-fly and can cause very different clinical manifestations depending on the infecting species and the host immune system (101). The disease is endemic in 88 countries and each year 1.6 million people are infected while approximately 350 million are at risk. There are at least four treatment options currently available including antimonials, amphotericin B, miltefosine and paromomycin, none of which are adapted to the field. The main drawbacks to these treatments are toxicity, long treatment courses, need for hospitalization, high costs, and resistance. The most promising candidates in the discovery and development pipeline rely on combination therapies. A high-throughput, high-content (image-based) cell-based assay using human macrophage cell lines infected with intracellular Leishmania sp. has also been developed (85,102,103,104,105,106,107,108,109,110,111,112,113,114,115).
Nuhs et al. developed and experimentally validated a novel axenic Leishmania in vitro assay that specifically can be used to identify cytocidal compounds with less false-positives than other assays (116). Using this novel assay, they screened 15,667 compounds in the primary screen at 15 μM and found 138 active hits and 15,529 inactive compounds. The 138 hits were then compared to results with intracellular assays, and 67 were shown to be active (49% of hits) (116). The hits that were inactive intracellularly may suggest that the targets are not essential intracellularly. Their results demonstrated that the new assay could robustly predict the intracellular amastigote Leishmania stage, which is currently the gold standard for in vitro drug screening (116).
De Rycker’s 2013 description of their assay optimization for Leishmania also resulted in hit compounds from two parallel studies. These authors performed an amastigote axenic assay that was more readily adaptable for high throughput but it did not differentiate between compounds that killed the parasite vs those that slow their growth (77). The authors screened almost 16,000 compounds and those that showed activity toward the amastigotes failed to perform at all or to the same extent in the more physiologically relevant intramacrophage assay the authors developed. 6 hits were selective for Leishmania as compared to human lung cells and exhibited dose-response relationships for intracellular Leishmania (77).
Sanderson et. al. used both in vitro and in vivo methodologies and found several kinase inhibitors as well as other compounds (sorafenib, sunitinib, lapatinib, PP2, amphotericin and miltefosine) used for cancer therapy to also be effective against Leishmania (71).
Multiple Kinetoplastids
Several studies have considered multiple kinetoplastids, such as the extensive work performed by Pena et al. (117). In order to identify potential new drugs, whole-cell phenotypic assays against Leishmania donovani, Trypanosoma cruzi, and Trypanosoma brucei were used to screen the GlaxoSmithKline HTS diversity set of 1.8 million compounds (117). For each of the three screens used, an orthogonal assay was conducted to ensure compounds with genuine activity were identified and enable compounds that interfered with the assays to be ruled out. The selection of candidates started first with the most potent, specific, and non-cytotoxic compounds in the dose-response outputs of each screen that was also filtered for lead-like compounds. The final boxes contained 592 compound: 192 of these compounds were active against Leishmania donovani, 222 were active against Trypanosoma cruzi, and 192 were active against Trypanosoma brucei. Kinases were the most frequent target class of the selected inhibitors. The compounds in the boxes mostly comprised of heterocyclic moieties that can bind cytochrome metal groups, nitro-substituted aryl groups, including nitro-pyrazoles, nitro-triazoles, nitro-furans, nitro-thiophenes, and nitro-benzenes (117).
Khare et al. screened 3 million compounds against the three kinetoplastids and identified a number of compounds with activities at 10 μM and with a selectivity index greater than 5 (26). They also synthesized 3,000 compounds for further optimization and produced an azabenzoxazole compound that was very active against all three species (26).
Some work has also been performed for multiple kinetoplastids using the MMV malaria box (55). Van Voorhis et al., reviewed 236 screens performed by many different groups that used the malaria box for a variety of repurposing efforts (55). Kaiser et al. performed a handful of these studies. They screened 400 compounds from the malaria box against bloodstream T. b. rhodesiense and T. brucei and against intracellular amastigotes of T. cruzi and L. infantum (118). Out of 400 compounds, 4 passed initial in vitro screening, but failed in in vivo testing (118). The same year, Kaiser et al took 100 drugs with a high likelihood of repurposing and tested them against several neglected tropical disease parasites. They tested some of the more potent compounds in mouse models for the various diseases with mixed results (33). For activity against T. b. rhodesiense they show the antidepressants sertraline and paroxetine to be good repurposing candidates. Several were reported for the first time for having moderate anti-Chagas efficacy such as tadalafil, an erectile dysfunction drug, and mebeverine, an antispasmodic (33). Promazine and nortriptyline were also identified in this study as anti-malarial compounds alongside several tricyclic antidepressants (33).
Schistosomiasis
Schistosomiasis is caused by a blood fluke (flatworm) and infects approximately 350 million people worldwide (119). The parasite is found in freshwater harboring the appropriate vector snails, and can actively invade skin. Children who play in water are particularly at risk (120). Once mature, the male and female flukes mate and lay eggs. These elicit a variety of immuno-inflammatory responses that can lead to a lifetime of pain and malaise (121), which interfere with school attendance and the ability to work, and consequently negatively impact both personal well-being and community development (122,123,124). Schistosomiasis also increases the risk of acquiring HIV infection (125,126,127).
Treatment and control of schistosomiasis relies on mass drug administration (MDA) with just one drug, praziquantel, which was developed 40 years ago (128,129,130,131). Apart from the obvious worry of drug resistance should PZQ fail, particularly as its use will being dramatically expanded in the next decade, the drug has a number of pharmaceutical and pharmacological weaknesses that encourage the search for a better chemotherapy (132). However, unlike the situation for malaria, other protozoal diseases and even the soil-transmitted helminthes (STHs) discussed here, there is no trans-nationally organized discovery or development program to find a better drug and little evidence that this situation will change anytime soon. Therefore, much if not all of the effort to identify new anti-schistosomal drugs occurs in the academic setting.
In the search for new chemical matter, the last decade has witnessed a number of advances in whole-organism (phenotypic) screening paradigms, which include the use viability- or vitality-based singe-metric assays (reviewed in (133)), and partially or fully-quantitative image analysis approaches (134,135,136,137,138). Regardless of format, most of the assays have focused on post-infective larvae (schistosomula), which are a relatively abundant stage of the parasite (Schistosoma mansoni being the ‘model’ schistosome) allowing for a reasonable screening throughput. The use of larval parasites is rationalized on the basis that the target product profile (TPP) for new anti-schistosomal drugs should encompass activity against immature schistosomes and not just mature parasites, as is the case with PZQ (139,140,141,142,143,144). Hits identified against schistosomula are then screened in vitro for activity against the more limiting adult parasites (which must be harvested from small animals such as mice or hamsters) before being then considered for in vivo efficacy studies (134,145). This staged or tiered approach has involved the screening of both chemically diverse and focused chemical collections and has identified a variety of anti-schistosomal chemistries, including antibiotics (e.g., doramectin and clofazimine (34)), neuroactives (134), anti-cancer agents (e.g., trametinib and vandetanib (108)) protease inhibitors (134), thiosemicarbazones (146) and statin drugs used for hypercholesterolemia (147). A few studies may be highlighted.
Mansour et al. obtained compounds from multiple libraries, i.e., from Pfizer and GSK, and performed high-throughput screening of 300,000 compounds on schistosomula followed by hit-screening against juvenile and mature parasites (28). They eventually prioritized seven compounds for future study, none of these were already approved drugs.
Ingram-Sieber et al. studied the antischistosomal properties of the MMV malaria box comprising 200 diverse drug-like and 200 probe-like compounds with confirmed in vitro activity against Plasmodium falciparum (malaria) (145). Five compounds were identified as lead compounds, and compound 2, a N,N9-diarylurea (IC50 = 0.83 μM), and compound 17, a 2,3-dianilinoquinoxaline (IC50 = 0.83 μM) were the most promising compounds to focus on for future antischistosomal studies. In vivo studies with mice infected with Schistosoma mansoni showed that a single oral dose of 400 mg/kg of the two drugs reduced worm burden by 52.5% (N,N9-diarylurea) and 40.8% (2,3-dianilinoquinoxaline). These two drugs represent a useful scaffold for preparing future small molecule inhibitors of S. mansoni (145). Apart from whole-organism screening, target–based approaches to defining novel anti-schistosomals have been employed. Liu et al. identified multiple compounds for repurposing for schistosomiasis by performing a high throughput docking screen of 14,000 molecules against an enzyme (3-oxoacyl-ACP reductase) involved in fatty acid synthesis. Compounds were tested directly for inhibition in a worm study, and for cell toxicity, resulting in 2 final compounds that both impacted mortality rates while exhibiting low cytotoxicity in human cells (40).
HDAC8, an enzyme involved in the epigenetic functions of Schistosoma mansoni was selected as a target for high throughput screening in another study (148). Through homology modeling Kannan et al. were able to perform docking of ~5000 compounds from classes with known zinc binding capabilities. From this screen, 75 compounds were selected for in vitro testing. 8 compounds showed good toxicity against the parasitic enzyme and are considered to be lead compounds (148).
Li et al. implemented a high throughput screen against 59,360 synthetic compounds targeting Schistosoma mansoni thioredoxin glutathione reductase (SmTGR), which is a well-characterized drug target for Schistosoma mansoni (25,149). Of the compounds, 74 showed a concentration-dependent inhibitory trend against thioredoxin glutathione reductase. Testing in cultured larvae showed that 39 compounds had cidal activity within 48 hours, while three of the compounds killed adult worms ex vivo at concentrations between 5 μM and 10 μM (25).
Targeting the same enzyme, using an integrated approach, Neves et al developed QSAR models for SmTGR and applied them to virtual screen a commercial database of 150,000 compounds (150). The computational approach allowed for the selection of 29 virtual hits, which were tested using high content screening assays against schistosomula and adult worms. Six compounds were active against schistosomula and three active against adult worms (hit rate of 20.6%). Among them, 2-[2-(3-methyl-4-nitro-5-isoxazolyl)vinyl]pyridine and 2-(benzylsulfonyl)-1,3-benzothiazole, two compounds representing new chemical scaffolds, showed inhibitory effect equivalent to PZQ, with EC50 values around 2.50 μM against both schistosomula and adult worms, representing promising new antischistosomal hits for further hit-to-lead optimization (150).
A composite approach, involving both whole-organism and target-based strategies, was recently employed to discover and validate a phosphodiesterase 4 (PDE4) as a schistosome drug target using benzoxaborole inhibitors of human PDE4 (151). Notably, the study employed a transgenic line of C. elegans that expressed the schistosome PDE4 in order to functionally characterize and validate the target.
Finally, chemogenomics and cheminformatic strategies have been employed to identify possible novel targets from anti-schistosomal drugs discovery. Neves et al. considered 2,114 proteins with the concept that "similar targets have similar drugs" and used each protein to search through 3 drug databases to find drugs with activities against Schistosoma flatworms (152). In addition to finding several compounds with confirmed positive activities, they suggested 115 untested compounds for potential repurposing (152). In the following study, the same authors (153) tested some of the suggested drugs in the previous study, in schistosomula and adult S. mansoni worms using a spectrophotometric assay and an automated image-based assay to accurately measure parasite viability and motility, respectively. They found that paroxetine, an antidepressant drug, showed a pronounced effect on schistosomula viability (IC50 =2.5 μM) after 72 h of incubation and EC50 values for motility reduction in male and female worms were 5.1 μM and 9.9 μM after 24 h of exposure, respectively (153).
Nematodes
Burns et al. used C. elegans as a model system for parasitic nematodes that are otherwise difficult to screen and identified 275 compounds that were lethal (154). They re-screened hits on two parasitic nematodes that infect livestock and two vertebrate models as a way of determining a degree of selectivity. Interestingly, they noted that nematicidal compounds have a higher logP (octanol-water partition coefficient) than the rest of the 67,012 compounds screened (154).
Mathew et al., took a similar approach screening 25,986 compounds from multiple libraries initially identifying 137 compounds active against C. elegans and focused on 14 compounds that impact both growth and fecundity (155). They grouped the compounds according to Tanimoto similarity. They then identified 9 compounds that fell into 2 clusters from the previously described Burns (154) study and 5 compounds with structural features not belonging to previously suggested nematocides. They compared activity of these compounds on the representative nematode to mammalian and yeast cells. Structure-activity relationships (SAR) and comparative genome sequencing were considered to gain an understanding of potential risk for resistance development. They described the drug discovery process as being inherently difficult to find compounds that are effective and present variety in mechanism of action which is important for combating resistance but in turn requires large libraries to be screened (155).
Crowther et al. also recognized the need for large screens for parasitic nematodes like Brugia malayi, the causative agent of lymphatic filariasis (156). Lymphatic filariasis and River blindness are two related diseases that together affect over 100 million people and are associated with debilitating symptoms (156). These neglected diseases stand to benefit from broad-spectrum nematocide screens described above and/or more target specific screens. Unfortunately, after screening almost 400,000 compounds through a C. elegans containing workflow they identified only 2 compounds effective against Brugia malayi but not at particularly low concentrations, which resulted in a discussion of the druggability of protein targets (156). The paradox discussed was over the need for a quality protein target worth the expenditure of a high throughput screen, and the difficulty in determining a protein’s promise before resources are spent. For this reason, some groups prefer to use smaller, enriched libraries of compounds.
Bilsland et al. developed an assay expressing genes for target proteins (both the worm and human counterpart) of the Brugia malayi parasite in yeast (157). They were then able to screen 400 compounds from the malaria box and found several selective compounds that were then tested in vitro. Several compounds killed the adult parasites while others impacted growth or motility in a significant manner. This is important since the majority of the symptoms of the disease are associated with the parasite in its adult form, there is a lack of drugs that function in this stage, and the initial screen improves the likelihood that these compounds are able to be taken up into a living organism (157).
Considering multiple filariid nematodes, Bulman et al. screened an FDA-approved library with adult worms of Brugia spp. (microfilariae) and Onchocerca onchengi, third-stage larvae (L3s) of Onchocerca volvulus, and the microfilariae of Onchocerca ochengi and Loa loa (47). Drug candidates were first selected by using the Worminator assay with adult female Brugia, which measures adult worm motility in response to drug. Compounds that inhibited motility by 75% were then screened against O. volvulus molting larvae and O. ochengi adult worms in MTT assay and motility assay. Bulman et al. found that auranofin, a gold containing compound, that was first developed to treat rheumatoid arthritis, was effective at killing adult Brugia and O. ochengi worms and in inhibiting the larval O. volvulus from molting from third-stage larvae to fourth-stage larvae (47). Auranofin was next tested in vivo in gerbils infected with B. pahangi L3 by measuring the burden of Brugia adult worms after treatment for 28 days at 5 mg/kg. Gold plasma levels were also measured in the gerbils to assess the circulation time of the drug. Based on previous studies, Bulman et al. hypothesized that the mechanism of auranofin inhibition might be due to auranofin targeting thioredoxin reductase and thioredoxin glutathione reductase (TGR) (47).
Stadelmann et al. evaluated the in vitro efficacy of the MMV malaria box, for the treatment of Echinococcus multilocularis, the parasitic worm that causes alveolar echinococcosis, the most deadly of helminth infections (158). The authors first utilized PGI assays, which measure the levels of enzyme phosphoglucose isomerase (PGI) release after damage, to narrow down the Malaria Box to 24 compounds that demonstrated physical damage in metacestodes (proliferative form of Echinococcus multilocularis). However, only seven of the twenty-four compounds were active at 1 μM (158). A dose-response assay demonstrated that only two of the seven compounds had EC50 values below 5 μM. The most active drugs from the PGI screen were also assessed in vitro for toxicity in human foreskin fibroblasts (HFF) and Reuber rat hepatoma (RH) cells. Cells were seeded in 96-well plates and 10 μM of each compound was added after 24 hours. After five days, the viability of the cells was assessed. The cascade of in vitro screening described led to the identification of compound MMV665807 as a potential treatment for Echinococcus multilocularis infections. However, the results could not be reproduced in vivo testing in mouse models (158).
Johnston et al. screened approximately 2,600 drugs approved for use in humans in an assay against Wolbachia, an endosymbiotic bacteria with filarial nematodes (42). Of the drugs screened, 69 hits were orally available, fifteen were selectively screened, and minocycline, methacycline, sparfloxacin (antibiotics), and rifapentine (anti-TB) were active in mouse models (42).
Protists and bacteria
Love et al. used a whole-cell phenotypic screening platform to test a library of 78,942 small molecules against Cryptosporidium parvum (41). This is a cause of diarrhea, which is associated with significant mortality in developing countries. Two libraries of compounds were used for this study namely a bioactive (10,000 compound) set assembled by the California Institute for Biomedical Research (Calibr) and the Global Health Chemical Diversity Library (GHCDL; 69,000) that was obtained from the University of Dundee Drug Discovery Unit. In vitro cultivation of Cryptosporidium has not yet been established, so in vivo oocyst propagation of C. parvum in calves was used in this study. The five most potent compounds against C. parvum were found to be Go ̈6976 (EC50 = 2.5 nM), monensin (EC50 = 7 nM), clofazimine (EC50 = 15 nM), cyclosporine (EC50 = 48 nM), and MST-312 (EC50 = 61 nM) (41). Go ̈6976 is known as a potent inhibitor of protein kinase C and of the tyrosine kinases JAK 2 and FLT3. MST-312 is also known as Telomerase Inhibitor IX and is a potent and reversible inhibitor of telomerase activity, arresting cells in the G0-G1 phase during the cell cycle. Cyclosporine is relatively old drug that is an inhibitor of the phosphatase activity of calcineurin and has well known potent immunosuppressive properties. Monensin is an inonophore antibiotic that is added to cattle feed, and finally clofazimine is an FDA-approved drug used for the treatment of leprosy that has recently also been evaluated for tuberculosis. However, only clofazimine showed a promising safety profile. Future work is still needed to establish whether clofazimine is targeting a parasitic pathway or modulating a host-mediated pathway essential to the parasite proliferation (41).
Several libraries of previously FDA-approved medications were assessed for their ability to inhibit the growth of the bacteria Borrelia burgdoferi, the causative agent of Lyme Disease in North America. From these studies, 150 compounds were identified and the top 20 further characterized for activity (159).
A standard phenotypic screen was performed using the Malaria box on two different protists (160). Toxoplasmosis is a widely spread disease which can be fatal to developing fetuses and individuals with weakened immune systems (161). Entamoeba histolytica is a disease especially pronounced in sub-Saharan Africa with mortality rates near that of malaria. For toxoplasmosis, seven compounds were identified that exhibited IC50’s of < 5μM and had a selectivity index of greater than 6. These compounds were compared to currently used anti-toxoplasmosis drugs, and it was determined that there was no overlap in scaffold type. This indicates the structural novelty of these compounds for this particular repurposing effort. This is beneficial for the cases in which drug-resistance has emerged. Two compounds were similarly identified for repurposing for Entamoeba histolytica (160).
Viruses
Dengue virus
Dengue is a vector-borne disease that causes 20,000 deaths a year and places nearly half the world’s population at risk (162). The presence of multiple serotypes is a challenge for successful anti-viral strategies as it places our own immune strategies at a disadvantage. The disease can progress to dengue hemorrhagic fever, also called severe dengue, which is most likely to occur in secondary infections and can be potentially fatal (163). A 2013 review by Lim et al. summarized some important work concerning repurposing for dengue (164) and discusses two strategies used for dengue drug therapy namely, inhibiting viral targets or inhibiting host targets. In recent work by Cruz et al, cell-based screening of 4,000 small molecules was performed on 4 different serotypes of the dengue virus in host cells resulting in 157 active compounds with 4 general structural scaffolds (162). If compounds were toxic to host cells the virus would die and this test would not distinguish between the two effects. Several scaffolds of compounds were identified as being toxic to multiple sources of the dengue virus. With the use of confocal microscopy and time-of-addition assays further insight was gained into the potential mechanism of action of broad-spectrum hits. It was determined that 2-aminothiazole was not interfering with viral entry, but was likely blocking virus assembly. This was hypothesized based on structural similarity to the more-well studied dasatinib (162).
In a high throughput screen, Yang et al., screened 60,000 compounds at a single concentration of 10 μM, and identified a potential small-molecule inhibitor of dengue virus BP13944 (a quaternary ammonium salt) (165). BP13944 inhibited replication or viral RNA synthesis in all four serotypes but not Japanese encephalitis virus (JEV) without any apparent cytotoxicity (EC50=1.03 μM). A stable reporter-DENV replicon cell line was used to screen the potency of inhibitors. They also demonstrated that E66G substitution in the NS3 region causes resistance of the virus to BP13944, suggesting that the inhibitor targets NS3 protease to inhibit viral replication and RNA synthesis. Future work is needed to understand the mechanism by which BP13944 interacts with NS3 protein to inhibit DENV replication (165).
Cheung et al. conducted screening of the US drug library collection (Microsource, Discovery Systems Inc., Gaylordsville, CT, USA), using an immunofluorescence based phenotypic screening assay (166). Lanatoside C, a cardiac glycoside, inhibited DENV-2 with an IC50 of 0.19 μM and low cytotoxicity (CC50 of 5.48 μM). The authors suggest that the compound inhibits the early processes of DENV replication cycle. Furthermore, after testing lanatoside C against the different DENV serotypes and other positive-strand RNA viruses, they found that it effectively inhibited all serotypes of DENV, flavivirus Kunjin, alphavirus Chikungunya and Sindbis virus as well as the human enterovirus (166)
A fluorescence-based high throughput assay, that tested DENV NS3 helicase activity, was used to screen a library of 1,600 compounds obtained from the Experimental Therapeutics Centre, Singapore (167). Among the compounds, suramin, a sleeping sickness drug, was shown to inhibit DENV helicase with an IC50 of 0.4 μM. The compound presented a noncompetitive mode of inhibition, with a Ki of 0.75 μM, indicating that it is able to bind to the apo form of the enzyme (167).
An immunofluorescence image-based assay identified the c-Src protein kinase inhibitors, dasatinib and AZD-0530 (saracatinib), as potent blockers of DENV (serotypes 1–4) and murine flavivirus Modoc (168). The Src-family kinases were hypothesized to have important roles in regulating DENV replication, based on the selectivity profiles of the compounds that exhibited anti-DENV activity in the primary and secondary screens. In this study they also screened a collection of 120 known inhibitors of mammalian Ser/Thr and Tyr kinases (168).
Zika virus
The Zika virus (ZIKV) is well known as an arthropod-borne flavivirus of the family Flaviviridae. It is phylogenetically close to dengue virus and yellow fever and is transmitted by Aedes mosquitoes, (169). It usually causes a mild dengue-like illness with possibly fever, joint pains, rash, and/or swollen lymph nodes (170). The virus has been associated in several studies with rare Guillain-Barré syndrome (171) and other severe neurological disorders (172). In 2015 ZIKV emerged due to an outbreak and was a major public health threat to the Americas (173). The World Health Organization deemed the cluster of microcephaly cases that were identified as well as the incidence of Guillain-Barré and other neurological disorders as all being associated with ZIKV in Latin America and the Caribbean. The WHO classified Zika as constituting a Public Health Emergency of International Concern (174) (PHEIC). We (175) and others (176,177,178) described steps that could be taken to initiate a drug discovery program on ZIKV including computational repurposing. We also described using the World Community Grid project called OpenZika to assist in antiviral drug discovery (179,180).
Several approaches to repurposing drugs for the Zika virus have been attempted in recent years to respond to the recent outbreak. Barrows et al., screened 774 FDA approved drugs for anti-Zika activity and several were selected to extend the screening to multiple types of human tissue relevant to Zika survival and transmission (45). Importantly, the authors also considered the relative risk of these identified compounds if they were to be used during pregnancy. Of the 13 candidate drugs only three fell into pregnancy category B, described as when “Animal reproduction studies have failed to demonstrate a risk to the fetus and there are no adequate and well-controlled studies in pregnant women.” The three compounds in this category were daptomycin, mefloquine HCl, and palonosetron HCl. Daptomycin is an anti-microbial, mefloquine HCl is an anti-parasitic that disrupts autophagy, and palonosetron HCl is a 5-hydroxytryptamine-3 receptor antagonist with drug interactions with SSRIs. All other compounds identified for anti-Zika activity were in the C and D pregnancy categories, where adverse effects have been observed in either humans or animals (45).
Pascoalino et al., developed, validated and utilized an assay against the Zika virus (32). They screened the NIH Clinical Collection and found 5 structurally diverse, selective anti-Zika compounds. These compounds were lovastatin, a hypolipidemic agent; 5-fluorouracil, an anti-cancer agent, 6-azauridine, a broad spectrum antimetabolite; palomosetron, a 5-HT antagonist; and kitasamycin, a macrolide, broad spectrum antibiotic (32).
Xu et al., had observed increased caspase-3 activity in Zika infected cells. They used this information to develop an assay and screen multiple libraries with a combined ~6,000 compounds (181). Initially, just over 100 compounds were identified as hits. Of these, emricasan, PHA-690509 and niclosamide showed significant activity without cytotoxicity. These were studied in combination and additive behavior was observed (181).
Retallack et al. performed a High-Throughput screen of 2,177 FDA-approved drugs, with an emphasis on drugs known to be safe in pregnancy (182). The screening was performed by monitoring inhibition of virus-dependent cell death in Vero or U87 cells. The authors showed that the antibiotic, azithromycin, reduces viral proliferation in glial cells, with low toxicity (182).
Ebola virus
The outbreak and spread of the Ebola virus (EBOV) in West Africa in 2014 highlighted the clear need for new antiviral drugs for this and other similar emerging viruses (183). A few research groups had previously undertaken high throughput screens and identified several already approved drugs with demonstrated in vitro growth inhibitory activities against the EBOV (48,184). A study by Kouznetsova et. al. used an assay that identifies compounds that block Ebola VLP (Virus-Like Particles) entry into host cells. From the 600 FDA-approved drugs that were screened 23 compounds were identified and the most potent group were microtubule inhibitors (44). The same group then identified 30 more hits from a larger library of over 2,000 compounds, and the authors went on to propose screening more from a 400,000 NCATS library (44).
In 2013, using an assay developed previously (185), Johansen et al., screened Food and Drug Administration (FDA)– and ex–US- approved drugs and molecular probes against an engineered strain and identified several compounds with activity against Ebola Zaire virus (51). Clomiphene, toremifene, tamoxifen, and raloxifene were particularly effective estrogen receptor antagonists along with diethylstilbestrol to a lesser extent. Quinestrol, equilin, and hydroxyprogesterone caproate were ER agonists and also provided antiviral activity. Extensive work was performed with multiple cell lines to investigate selectivity (51). Two years later, this same group screened a library of 2600 US and non-US approved drugs through an in vitro high throughput screen of Ebola. Eighty compounds were considered initial hits, 30 were selected for further screening and eventually 4 top compounds were confirmed in mouse efficacy studies (50).
Edwards et al., optimized an Ebola minigenome assay and tested a library of over 2000 compounds with the transfected cells and then moved on to test 5 compounds under biosafety level 4 conditions with infectious virus (35). Mycophenolic acid and gedunin showed quite promising results with significantly reduced virus replication and low cytotoxicity (35).
In an exhaustive study performed by Anantpadma et al., 319,855 small molecules were screened at high concentration against Ebola and Marburg Virus Like Particles transduced into Hela cells (31). There were several steps to the screen which reduced final hits to 17 compounds that are active against both viruses (31).
Cheng et al., transfected human kidney cells with plasmids from multiple virus types including Marburg, EBOV, and HIV creating pseudovirions for their high throughput assay (186). They investigated the Prestwick Chemical Library and discovered 20 compounds that were effective against both Marburg and EBOV. A striking number of hits were GPCR (G-Protein Coupled Receptor) antagonists which provided additional insight to potential mechanism of action (186). Specifically, time of addition studies indicated that the inhibitors block viral entry after initial attachment and before cell fusion either by direct antagonism, indirectly via GPCR initiated cell signaling, or a combination. The compounds identified in this study were diverse in structure and target. Histamine receptors, 5-HT (serotonin) receptors, muscarinic acetylcholine receptors, adrenergic receptors were found to block viral entry. Clozapine and trimipramine maleate salt fell into all of the above listed categories and 4 compounds were listed as having “other targets.” Benztropine displayed broad spectrum antifilovirus activity against multiple isolates of EBOV and MARV (186).
Wang et al. performed a screen of 1280 FDA-approved drugs using EBOV (the Zaire strain of Ebola virus) GP/HIV core pseudovirus that contains firefly luciferase reporter gene in order to identify new targets for the treatment of Ebola (46). Of the 1280 compounds, 137 compounds had greater than 50% inhibition of pEBOV at 10 μM. Of the 137 compounds, 15 compounds that had less than 15% inhibition of vesicular stomatitis virus (pVSV) were identified as selective primary hits. To validate the 15 primary hits, the cytotoxicity of each compound was assessed in parallel without the pseudovirus, this was necessary to rule out compounds that inhibited FLuc expression or HIV replication. Two compounds were identified as hits including teicoplanin which is a glycopeptide antibiotic used for the treatment of gram-positive bacterial infections (EC50 2.38 μM) and toremiphene (EC50 0.38 μM). Teicoplanin was selected for further characterization, and demonstrated complete inhibition of EBOV without apparent cytotoxity, making it an ideal candidate for the treatment of Ebola (46).
Bayesian machine learning models were developed using data sets from previous drug screening against the Ebola virus (48). This data was used to generate Bayesian models that were used to score the MicroSource Spectrum library to predict compounds that would display anti-Ebola activity. Quinacrine, pyronaridine, and tilorone were identified and their activities were successfully confirmed in vitro as having good potency (187). Most recently, tilorone was shown to have 100% efficacy in a mouse model of Ebola infection (188).
Mirza et al. considered two target proteins of the EBOV-Z Virus and virtually screened 45,013 compounds after filtering compounds from a much larger compilation of libraries including phytochemical compounds and natural products (189). The proteins for which crystal structures were available were energy minimized using CHARMM and proteins for strains of Ebola without resolved crystal structures were homology modeled and energy minimized for docking. 749 hits were considered based on their predicted ADMET properties and 13 were selected for further study. The authors performed retrospective analysis, with known in vitro knowledge from the literature and aggregation prediction to account for what would likely be false positives in an in vitro assay. To date there has not been any in vitro validation (189).
Zhao et al., used a homology model for VP24, an enzyme critical to Ebola-human interaction and used molecular dynamics to find a conformation (49). Once this was found, they screened all FDA approved drugs and 259 nucleotide/nucleoside drugs and investigated docked poses. Multiple docking programs were compared and 15 compounds were finally selected as potentially having activity that should be tested in vitro and in vivo against Ebola (49).
Chopra et al. conducted a high throughput screen of compounds with known side effects including FDA approved drugs through almost 50,000 protein structures (190). The interpretation of the resulting drug-protein interactions led to 46 compounds that had been previously established as having activity against Ebola by previous in vitro work. 8 novel suggestions were provided by this CANDO approach and are suggested for experimental follow up although this was not performed (190).
Other Viruses
Madrid et al. screened 1012 FDA-approved drugs at multiple dosages against multiple biological threat agents including Ebola, the Marburg virus, the Lassa virus, and Bacillus anthracis, Francisella tularensis, and Coxiella burnetii (causative agents for Anthrax, Tularemia, and Q fever respectively) (48). Initial inhibition was compared to positive controls (bafilomycin A1 for viruses, and ciproflaxin for bacteria) and compounds that performed comparably (within two standard deviations) were selected for further intracellular screens. The hits were further narrowed by selecting compounds with broad activities against more than one bacteria or virus (48). The Ebola dataset from this study was subsequently used in a machine learning model described earlier (187).
Mudhasani et al., investigated a library of protease inhibitors against the Rift Valley Fever Virus (RVFV) (191). 839 compounds from the ChemDiv library were screened and 34 compounds resulted in over 50% inhibition with four distinct structure types. Time of addition studies were also performed to gain insight into the mechanism of action, i.e. which compounds are involved with virus entry, viral RNA transcription inhibition, or protein synthesis disruption (191).
Cruz et al., performed similar work in which they screened a library of 4,000 kinase inhibitors against the Chikungunya Virus (192). They identified 76 compounds which provided greater than 50% inhibition. Of those, only 6 of those were dose dependent and were characterized by image-based screening (192).
Karlas et al. utilized a host genome-wide loss-of-function screen to validate 156 proviral and 41 antiviral host factors (193). Sixteen of these factors were relevant to multiple viruses and represented the central biological nodes for antiviral therapeutic intervention. To validate the sixteen factors, CRISPR/Cas9 technology was used to generate cells that were deficient in these genes and the functional response was observed. Gene enrichment analysis was used to analyze the biological processes and molecular functions that are required for viral infection. Karlas et al. next assessed the antiviral activity of chemical compounds that target the identified proviral factors involved in chikungunya replication (193). They utilized specialized drug databases to identify 52 compounds that interact with gene products of 14 distinct chikungunya proviral genes, and 20 compounds were shown to interact with six distinct proviral factors or pathways. RNAi (RNA interference) screens showed that five out of six proviral factors were druggable. Special knockout mice were generated to study these results in vivo, and it was demonstrated that tivozanib significantly reduced chikungunya viral load in vital organs. Pimozide, a calmodulin inhibitor and TOFA, a fatty acid synthesis inhibitor also showed promising results in vivo (193).
Dyall and coworkers suggested the use of Emetine dihydrochloride hydrate, an antibacterial agent, and several other compounds to combat MERS-CoV and SARS-CoV (194). The authors began with an exclusive library of approved drugs that had shown in vitro activity tested in relevant cell lines and virus strains consisting of 290 compounds and found 66 which had efficacy against one or both of the viruses tested (194).
Molecular property analysis
We are not aware of others performing analysis across compounds identified by repurposing screens. Therefore we have curated the molecules derived from these various drug repurposing efforts and calculated molecular properties for the compounds. Several molecular descriptors were generated for compounds using Discovery Studio version 4.1 (Biovia, San Diego, CA) and summary statistics performed using JMP (Cary, NC). Table III and Figure S1 shows that for over 1,225 compounds the mean properties are very similar to drug-like properties summarized previously (195). In particular, we find the average MW to be 394.39 +/- 190.17 g/mol (less than 500), the number of H-bond donors to be 1.69 +/- 2.71 (less than 5), H-bond acceptors to be 5.05 +/- 3.50 (less than 10) and the average octanol-water partition coefficient to be 3.12 +/- 1.84 (less than 5). Since the majority of these compounds were identified through screening of large libraries (bioactives, FDA approved drugs, etc.) this result is not entirely surprising.
Machine learning models
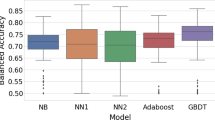
For several NTDs datasets we have developed Bayesian models using Assay Central (196,197,198) with datasets published by ourselves or others, for example Chagas disease, Ebola, Zika, etc (Figure 1). Several recent publications have described Assay Central in more detail as well as made comparisons with other algorithms (196,197,198). The NTD models can therefore be used to score input molecules and prioritize them for testing which is ongoing in our labs and those of collaborators. This work expands upon our previous efforts for Chagas (79), Ebola (199) and other diseases such as tuberculosis (196). We believe there are considerable opportunities for drug discovery that could be achieved by mining the growing public datasets for neglected diseases.

Examples of Bayesian models for (a) Chagas disease, (b) Dengue virus, (c) Ebola virus, (d) L. donovani, (e) L. infantum, (f) Toxoplasma gondii, developed with Assay Central.
Conclusions
The clear trend towards using phenotypic screens in place of target-based screens is particularly persuasive for NTDs, as well as for other bacterial, viral and fungal pathogens. For these diseases, it is certainly considered more difficult to convert a strong targeted hit into a cell permeable, non-toxic drug than it is to identify the target of a non-toxic compound with phenotypic, whole-cell activity (200). This is certainly true in the case of intracellular parasites in which the compound has to traverse one or more of the host membranes to reach its final target. This study indicates that many different research groups globally have performed considerable amounts of work to identify compounds for repurposing for neglected diseases. These efforts still do not cover all the diseases defined by the WHO (Table I). Due to space restrictions we have not covered all the diseases eligible for a priority review voucher (Table II) for which there have been repurposing efforts (e.g. tuberculosis, malaria, etc). The 1,225 compounds identified from the various studies described (Supplemental data 1) appear to have generally “drug-like” properties as one would expect (Table III).
In some cases when disease transmission is rampant and the risk begins to outweigh costs, certain government agencies and academic institutions are more likely to be granted funding to pursue treatments for these diseases, as happened with Ebola and Zika viruses. In some instances, academics are able to partner with companies or increasingly they have access to their own large libraries of compounds that they can screen, to produce hundreds or tens of compounds with high likelihood of repurposing. Some of these groups have the tools and access to perform in vivo work and suggest clinical studies as well. Other groups perform in vitro screening and suggest follow-up with in vivo studies. Those various researchers using computational tools such as, molecular dynamics, docking, and machine learning models can provide considerable insight to specific mechanisms, perform high throughput screening very quickly, and even leverage machine learning to predict novel structures. However, it is common for these scientists to conclude with suggestions for validation in vitro and in vivo. Ideally this should be performed at the same time. From our own experience of collaborations on Chagas disease we have been able to go from in silico to in vitro to in vivo in a single study (79) and then for Ebola we went from in silico to in vitro in one study (187) and in vivo efficacy study in another (188). Most groups generally illustrate that they can go from in vitro to in vivo.
Repurposing drugs for neglected diseases likely requires the successful integration of multiple fields; it also helps to be aware of promising compounds and conversely potentially difficult compounds. Occasionally we see the same compound having disparate activities. For example clofazimine as an anti-trypanosomal agent (34) and active against Cryptosporidium parvum (41), and pyronaridine active against Chagas disease (79) and Ebola (187) (Fig. 2). Fexinidazole, which recently completed clinical trials for HAT (201), is also being considered for treatment of Chagas (202,203). Such molecules that have multiple potential activities against neglected tropical diseases may be more common than we think and could point to common targets across diseases. Further screens like those described herein will help identify such compounds and bring more treatments into the pipelines for these diseases. We hope that this review will inspire others to search for new molecules for NTDs.

Examples of molecules that have multiple activities against NTDs. (a) clofazimine, (b) pyronaridine.
References
Kotz J. Phenotypic screening, take two. SciBX. 2012;5:15.
Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6(1):29–40.
Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10(7):507–19.
Hotez PJ, Molyneux DH, Fenwick A, Kumaresan J, Sachs SE, Sachs JD, et al. Control of neglected tropical diseases. N Engl J Med. 2007;357(10):1018–27.
WHO. Neglected tropical diseases. Available from: http://www.who.int/neglected_diseases/diseases/en/.
Hotez PJ, Pecoul B. "Manifesto" for advancing the control and elimination of neglected tropical diseases. PLoS Negl Trop Dis. 2010;4(5):e718.
Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465(7296):311–5.
Ribeiro I, Sevcsik AM, Alves F, Diap G, Don R, Harhay MO, et al. New, improved treatments for Chagas disease: from the R&D pipeline to the patients. PLoS Negl Trop Dis. 2009;3(7):e484.
Bettiol E, Samanovic M, Murkin AS, Raper J, Buckner F, Rodriguez A. Identification of three classes of heteroaromatic compounds with activity against intracellular Trypanosoma cruzi by chemical library screening. PLoS Negl Trop Dis. 2009;3(2):e384.
Ponder EL, Freundlich JS, Sarker M, Ekins S. Computational models for neglected diseases: gaps and opportunities. Pharm Res. 2014;31(2):271–7.
FDA. Tropical Disease Priority Review Vouchers Guidance for Industry. Available from: https://www.fda.gov/downloads/Drugs/Guidances/UCM080599.pdf.
Berman J, Radhakrishna T. The Tropical Disease Priority Review Voucher: A Game-Changer for Tropical Disease Products. Am J Trop Med Hyg. 2017;96(1):11–3.
Ridley DB. Priorities for the Priority Review Voucher. Am J Trop Med Hyg. 2017;96(1):14–5.
Gaffney A, Mezher M, Brennan Z. Regulatory Explainer: Everything You Need to Know About FDA’s Priority Review Vouchers Available from: https://www.raps.org/regulatory-focus/news-articles/2017/12/regulatory-explainer-everything-you-need-to-know-about-fdas-priority-review-vouchers.
Boguski MS, Mandl KD, Sukhatme VP. Drug discovery. Repurposing with a difference. Science. 2009;324(5933):1394–5.
Uliana SR, Barcinski MA. Repurposing for neglected diseases. Science. 2009;326(5955):935 author reply 935.
Huang R, Southall N, Wang Y, Yasgar A, Shinn P, Jadhav A, et al. The NCGC Pharmaceutical Collection: A Comprehensive Resource of Clinically Approved Drugs Enabling Repurposing and Chemical Genomics. Sci Transl Med. 2011;3(80):80ps16.
Kinnings SL, Liu N, Tonge PJ, Jackson RM, Xie L, Bourne PE. A machine learning-based method to improve docking scoring functions and its application to drug repurposing. J Chem Inf Model. 2011;51(2):408–19.
Oprea TI, Mestres J. Drug Repurposing: Far Beyond New Targets for Old Drugs. AAPS J. 2012;14:759–63.
Blatt J, Farag S, Corey SJ, Sarrimanolis Z, Muratov E, Fourches D, et al. Expanding the scope of drug repurposing in pediatrics: The Children's Pharmacy Collaborative. Drug Discov Today. 2014;19:1696–8.
Martorana A, Perricone U, Lauria A. The Repurposing of Old Drugs or Unsuccessful Lead Compounds by in Silico Approaches: New Advances and Perspectives. Curr Top Med Chem. 2016;16(19):2088–106.
Baker NC, Ekins S, Williams AJ, Tropsha A. A bibliometric review of drug repurposing. Drug Discov Today. 2018;23(3):661–72.
Dudley JT, Deshpande T, Butte AJ. Exploiting drug-disease relationships for computational drug repositioning. Brief Bioinform. 2011;12(4):303–11.
Dimasi JA. New drug development in the United States from 1963 to 1999. Clin Pharmacol Ther. 2001;69(5):286–96.
Li T, Ziniel PD, He PQ, Kommer VP, Crowther GJ, He M, et al. High-throughput screening against thioredoxin glutathione reductase identifies novel inhibitors with potential therapeutic value for schistosomiasis. Infect Dis Poverty. 2015;4:40.
Khare S, Nagle AS, Biggart A, Lai YH, Liang F, Davis LC, et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature. 2016;537(7619):229–33.
Diaz R, Luengo-Arratta SA, Seixas JD, Amata E, Devine W, Cordon-Obras C, et al. Identification and characterization of hundreds of potent and selective inhibitors of Trypanosoma brucei growth from a kinase-targeted library screening campaign. PLoS Negl Trop Dis. 2014;8(10):e3253.
Mansour NR, Paveley R, Gardner JM, Bell AS, Parkinson T, Bickle Q. High Throughput Screening Identifies Novel Lead Compounds with Activity against Larval, Juvenile and Adult Schistosoma mansoni. PLoS Negl Trop Dis. 2016;10(4):e0004659.
Cole ST. Tuberculosis drug discovery needs public-private consortia. Drug Discov Today. 2016;22:477–8.
Ekins S. A summary of some EU funded Tuberculosis drug discovery collaborations. Drug Discov Today. 2017;22(3):479–80.
Anantpadma M, Kouznetsova J, Wang H, Huang R, Kolokoltsov A, Guha R, et al. Large-Scale Screening and Identification of Novel Ebola Virus and Marburg Virus Entry Inhibitors. Antimicrob Agents Chemother. 2016;60(8):4471–81.
Pascoalino BS, Courtemanche G, Cordeiro MT, Gil LH, Freitas-Junior L. Zika antiviral chemotherapy: identification of drugs and promising starting points for drug discovery from an FDA-approved library. F1000Res. 2016;5:2523.
Kaiser M, Maser P, Tadoori LP, Ioset JR, Brun R. Antiprotozoal Activity Profiling of Approved Drugs: A Starting Point toward Drug Repositioning. PLoS One. 2015;10(8):e0135556.
Panic G, Vargas M, Scandale I, Keiser J. Activity Profile of an FDA-Approved Compound Library against Schistosoma mansoni. PLoS Negl Trop Dis. 2015;9(7):e0003962.
Edwards MR, Pietzsch C, Vausselin T, Shaw ML, Bukreyev A, Basler CF. High-Throughput Minigenome System for Identifying Small-Molecule Inhibitors of Ebola Virus Replication. ACS Infect Dis. 2015;1(8):380–7.
Planer JD, Hulverson MA, Arif JA, Ranade RM, Don R, Buckner FS. Synergy testing of FDA-approved drugs identifies potent drug combinations against Trypanosoma cruzi. PLoS Negl Trop Dis. 2014;8(7):e2977.
Xu M, Lee EM, Wen Z, Cheng Y, Huang WK, Qian X, et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat Med. 2016.
Cheng H, Lear-Rooney CM, Johansen L, Varhegyi E, Chen ZW, Olinger GG, et al. Inhibition of Ebola and Marburg viral entry by G protein-coupled receptor antagonists. J Virol. 2015.
Keenan M, Alexander PW, Chaplin JH, Abbott MJ, Diao H, Wang Z, et al. Selection and optimization of hits from a high-throughput phenotypic screen against Trypanosoma cruzi. Future Med Chem. 2013;5(15):1733–52.
Liu J, Dyer D, Wang J, Wang S, Du X, Xu B, et al. 3-oxoacyl-ACP reductase from Schistosoma japonicum: integrated in silico-in vitro strategy for discovering antischistosomal lead compounds. PLoS One. 2013;8(6):e64984.
Love MS, Beasley FC, Jumani RS, Wright TM, Chatterjee AK, Huston CD, et al. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Negl Trop Dis, e0005373. 2017;11(2).
Johnston KL, Ford L, Umareddy I, Townson S, Specht S, Pfarr K, et al. Repurposing of approved drugs from the human pharmacopoeia to target Wolbachia endosymbionts of onchocerciasis and lymphatic filariasis. Int J Parasitol Drugs Drug Resist. 2014;4(3):278–86.
Gunatilleke SS, Calvet CM, Johnston JB, Chen CK, Erenburg G, Gut J, et al. Diverse inhibitor chemotypes targeting Trypanosoma cruzi CYP51. PLoS Negl Trop Dis. 2012;6(7):e1736.
Kouznetsova J, Sun W, Martinez-Romero C, Tawa G, Shinn P, Chen CZ, et al. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg Microbes Infect. 2014;3(12):e84.
Barrows NJ, Campos RK, Powell ST, Prasanth KR, Schott-Lerner G, Soto-Acosta R, et al. A Screen of FDA-Approved Drugs for Inhibitors of Zika Virus Infection. Cell Host Microbe. 2016;20:259–70.
Wang Y, Cui R, Li G, Gao Q, Yuan S, Altmeyer R, et al. Teicoplanin inhibits Ebola pseudovirus infection in cell culture. Antivir Res. 2016;125:1–7.
Bulman CA, Bidlow CM, Lustigman S, Cho-Ngwa F, Williams D, Rascon AA Jr, et al. Repurposing auranofin as a lead candidate for treatment of lymphatic filariasis and onchocerciasis. PLoS Negl Trop Dis. 2015;9(2):e0003534.
Madrid PB, Chopra S, Manger ID, Gilfillan L, Keepers TR, Shurtleff AC, et al. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS One. 2013;8(4):e60579.
Zhao Z, Martin C, Fan R, Bourne PE, Xie L. Drug repurposing to target Ebola virus replication and virulence using structural systems pharmacology. BMC Bioinformatics. 2016;17:90.
Johansen LM, DeWald LE, Shoemaker CJ, Hoffstrom BG, Lear-Rooney CM, Stossel A, et al. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci Transl Med. 2015;7(290):290ra289.
Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, et al. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med. 2013;5(190):190ra179.
Organization WH. Unprecendented progress against neglected tropical diseases. Available from: http://www.who.int/en/news-room/detail/19-04-2017-unprecedented-progress-against-neglected-tropical-diseases-who-reports.
Hotez PJ. Ten failings in global neglected tropical diseases control. PLoS Negl Trop Dis. 2017;11(12):e0005896.
Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol. 2014;32(1):40–51.
Van Voorhis WC, Adams JH, Adelfio R, Ahyong V, Akabas MH, Alano P, et al. Open source drug discovery with the malaria box compound collection for neglected diseases and beyond. PLoS Pathog. 2016;12(7):e1005763.
Manzano M, Padia J, Padmanabhan R. Small molecule inhibitor discovery for dengue virus protease using high-throughput screening. Methods Mol Biol. 2014;1138:331–44.
Uebelhoer LS, Albarino CG, McMullan LK, Chakrabarti AK, Vincent JP, Nichol ST, et al. High-throughput, luciferase-based reverse genetics systems for identifying inhibitors of Marburg and Ebola viruses. Antivir Res. 2014;106:86–94.
Alonso-Padilla J, Cotillo I, Presa JL, Cantizani J, Pena I, Bardera AI, et al. Automated high-content assay for compounds selectively toxic to Trypanosoma cruzi in a myoblastic cell line. PLoS Negl Trop Dis. 2015;9(1):e0003493.
Calvo-Alvarez E, Stamatakis K, Punzon C, Alvarez-Velilla R, Tejeria A, Escudero-Martinez JM, et al. Infrared fluorescent imaging as a potent tool for in vitro, ex vivo and in vivo models of visceral leishmaniasis. PLoS Negl Trop Dis. 2015;9(3):e0003666.
Paape D, Bell AS, Heal WP, Hutton JA, Leatherbarrow RJ, Tate EW, et al. Using a non-image-based medium-throughput assay for screening compounds targeting N-myristoylation in intracellular Leishmania amastigotes. PLoS Negl Trop Dis. 2014;8(12):e3363.
Zmurko J, Marques RE, Schols D, Verbeken E, Kaptein SJ, Neyts J. The Viral Polymerase Inhibitor 7-Deaza-2'-C-Methyladenosine Is a Potent Inhibitor of In Vitro Zika Virus Replication and Delays Disease Progression in a Robust Mouse Infection Model. PLoS Negl Trop Dis. 2016;10(5):e0004695.
Ochiana SO, Pandarinath V, Wang Z, Kapoor R, Ondrechen MJ, Ruben L, et al. The human Aurora kinase inhibitor danusertib is a lead compound for anti-trypanosomal drug discovery via target repurposing. Eur J Med Chem. 2013;62:777–84.
Patel G, Karver CE, Behera R, Guyett PJ, Sullenberger C, Edwards P, et al. Kinase scaffold repurposing for neglected disease drug discovery: discovery of an efficacious, lapatinib-derived lead compound for trypanosomiasis. J Med Chem. 2013;56(10):3820–32.
Guyett PJ, Behera R, Ogata Y, Pollastri M, Mensa-Wilmot K. Novel Effects of Lapatinib Revealed in the African Trypanosome by Using Hypothesis-Generating Proteomics and Chemical Biology Strategies. Antimicrob Agents Chemother. 2017;61(2).
Mhashilkar AS, Vankayala SL, Liu C, Kearns F, Mehrotra P, Tzertzinis G, et al. Identification of Ecdysone Hormone Receptor Agonists as a Therapeutic Approach for Treating Filarial Infections. PLoS Negl Trop Dis. 2016;10(6):e0004772.
Diaz-Gonzalez R, Kuhlmann FM, Galan-Rodriguez C, Madeira da Silva L, Saldivia M, Karver CE, et al. The susceptibility of trypanosomatid pathogens to PI3/mTOR kinase inhibitors affords a new opportunity for drug repurposing. PLoS Negl Trop Dis. 2011;5(8):e1297.
Hart CJ, Munro T, Andrews KT, Ryan JH, Riches AG, Skinner-Adams TS. A novel in vitro image-based assay identifies new drug leads for giardiasis. Int J Parasitol Drugs Drug Resist. 2017;7(1):83–9.
Lee H, Ren J, Nocadello S, Rice AJ, Ojeda I, Light S, et al. Identification of novel small molecule inhibitors against NS2B/NS3 serine protease from Zika virus. Antivir Res. 2017;139:49–58.
McCarthy SD, Majchrzak-Kita B, Racine T, Kozlowski HN, Baker DP, Hoenen T, et al. A Rapid Screening Assay Identifies Monotherapy with Interferon-ss and Combination Therapies with Nucleoside Analogs as Effective Inhibitors of Ebola Virus. PLoS Negl Trop Dis. 2016;10(1):e0004364.
Shahinas D, Debnath A, Benedict C, McKerrow JH, Pillai DR. Heat shock protein 90 inhibitors repurposed against Entamoeba histolytica. Front Microbiol. 2015;6:368.
Sanderson L, Yardley V, Croft SL. Activity of anti-cancer protein kinase inhibitors against Leishmania spp. J Antimicrob Chemother. 2014;69(7):1888–91.
Rassi A Jr, Rassi A, Marin-Neto JA. Chagas disease. Lancet. 2010;375(9723):1388–402.
Coura JR, Vinas PA. Chagas disease: a new worldwide challenge. Nature. 2010;465(7301):S6–7.
Campbell NCR, van Loon JA, Sundaram RS, Ames MM, Hansch C, Weinshilboum R. Human and rat liver phenol sulfotransferase: Structure-activity relationships for phenolic substrates. Mol Pharmacol. 1987;32:813–9.
Hotez PJ, Dumonteil E, Woc-Colburn L, Serpa JA, Bezek S, Edwards MS, et al. Chagas disease: "the new HIV/AIDS of the Americas". PLoS Negl Trop Dis. 2012;6(5):e1498.
De Rycker M, Thomas J, Riley J, Brough SJ, Miles TJ, Gray DW. Identification of Trypanocidal Activity for Known Clinical Compounds Using a New Trypanosoma cruzi Hit-Discovery Screening Cascade. PLoS Negl Trop Dis. 2016;10(4):e0004584.
De Rycker M, Hallyburton I, Thomas J, Campbell L, Wyllie S, Joshi D, et al. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrob Agents Chemother. 2013;57(7):2913–22.
Riley J, Brand S, Voice M, Caballero I, Calvo D, Read KD. Development of a Fluorescence-based Trypanosoma cruzi CYP51 Inhibition Assay for Effective Compound Triaging in Drug Discovery Programmes for Chagas Disease. PLoS Negl Trop Dis. 2015;9(9):e0004014.
Ekins S, Lage de Siqueira-Neto J, McCall L-I, Sarker M, Yadav M, Ponder EL, et al. Machine Learning Models and Pathway Genome Data Base for Trypanosoma cruzi Drug Discovery. PLoS Negl Trop Dis. 2015;9(6):e0003878.
Wiggers HJ, Rocha JR, Fernandes WB, Sesti-Costa R, Carneiro ZA, Cheleski J, et al. Non-peptidic cruzain inhibitors with trypanocidal activity discovered by virtual screening and in vitro assay. PLoS Negl Trop Dis. 2013;7(8):e2370.
Chatelain E. Chagas disease drug discovery: toward a new era. J Biomol Screen. 2015;20(1):22–35.
Molina I, Gomez i Prat J, Salvador F, Trevino B, Sulleiro E, Serre N, et al. Randomized trial of posaconazole and benznidazole for chronic Chagas' disease. N Engl J Med. 2014;370(20):1899–908.
Torrico F, Gascon J, Ortiz L, Alonso-Vega C, Pinazo MJ, Schijman A, et al. Treatment of adult chronic indeterminate Chagas disease with benznidazole and three E1224 dosing regimens: a proof-of-concept, randomised, placebo-controlled trial. Lancet Infect Dis. 2018;18(4):419–30.
Sutherland CS, Yukich J, Goeree R, Tediosi F. A literature review of economic evaluations for a neglected tropical disease: human African trypanosomiasis ("sleeping sickness"). PLoS Negl Trop Dis. 2015;9(2):e0003397.
McCall LI, McKerrow JH. Determinants of disease phenotype in trypanosomatid parasites. Trends Parasitol. 2014;30(7):342–9.
Mashiyama ST, Koupparis K, Caffrey CR, McKerrow JH, Babbitt PC. A global comparison of the human and T. brucei degradomes gives insights about possible parasite drug targets. PLoS Negl Trop Dis. 2012;6(12):e1942.
Navarro G, Chokpaiboon S, De Muylder G, Bray WM, Nisam SC, McKerrow JH, et al. Hit-to-lead development of the chamigrane endoperoxide merulin A for the treatment of African sleeping sickness. PLoS One. 2012;7(9):e46172.
Frevert U, Movila A, Nikolskaia OV, Raper J, Mackey ZB, Abdulla M, et al. Early invasion of brain parenchyma by African trypanosomes. PLoS One. 2012;7(8):e43913.
Mackey ZB, Koupparis K, Nishino M, McKerrow JH. High-throughput analysis of an RNAi library identifies novel kinase targets in Trypanosoma brucei. Chem Biol Drug Des. 2011;78(3):454–63.
Watts KR, Ratnam J, Ang KH, Tenney K, Compton JE, McKerrow J, et al. Assessing the trypanocidal potential of natural and semi-synthetic diketopiperazines from two deep water marine-derived fungi. Bioorg Med Chem. 2010;18(7):2566–74.
Mallari JP, Shelat AA, Obrien T, Caffrey CR, Kosinski A, Connelly M, et al. Development of potent purine-derived nitrile inhibitors of the trypanosomal protease TbcatB. J Med Chem. 2008;51(3):545–52.
Vicik R, Hoerr V, Glaser M, Schultheis M, Hansell E, McKerrow JH, et al. Aziridine-2,3-dicarboxylate inhibitors targeting the major cysteine protease of Trypanosoma brucei as lead trypanocidal agents. Bioorg Med Chem Lett. 2006;16(10):2753–7.
Mackey ZB, O'Brien TC, Greenbaum DC, Blank RB, McKerrow JH. A cathepsin B-like protease is required for host protein degradation in Trypanosoma brucei. J Biol Chem. 2004;279(46):48426–33.
Greenbaum DC, Mackey Z, Hansell E, Doyle P, Gut J, Caffrey CR, et al. Synthesis and structure-activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J Med Chem. 2004;47(12):3212–9.
Caffrey CR, Schanz M, Nkemngu NJ, Brush M, Hansell E, Cohen FE, et al. Screening of acyl hydrazide proteinase inhibitors for antiparasitic activity against Trypanosoma brucei. Int J Antimicrob Agents. 2002;19(3):227–31.
Caffrey CR, Hansell E, Lucas KD, Brinen LS, Alvarez Hernandez A, Cheng J, et al. Active site mapping, biochemical properties and subcellular localization of rhodesain, the major cysteine protease of Trypanosoma brucei rhodesiense. Mol Biochem Parasitol. 2001;118(1):61–73.
Troeberg L, Chen X, Flaherty TM, Morty RE, Cheng M, Hua H, et al. Chalcone, acyl hydrazide, and related amides kill cultured Trypanosoma brucei brucei. Mol Med. 2000;6(8):660–9.
Troeberg L, Morty RE, Pike RN, Lonsdale-Eccles JD, Palmer JT, McKerrow JH, et al. Cysteine proteinase inhibitors kill cultured bloodstream forms of Trypanosoma brucei brucei. Exp Parasitol. 1999;91(4):349–55.
Blaazer AR, Orrling KM, Shanmugham A, Jansen C, Maes L, Edink E, et al. Fragment-based screening in tandem with phenotypic screening provides novel antiparasitic hits. J Biomol Screen. 2015;20(1):131–40.
Bland ND, Wang C, Tallman C, Gustafson AE, Wang Z, Ashton TD, et al. Pharmacological validation of Trypanosoma brucei phosphodiesterases B1 and B2 as druggable targets for African sleeping sickness. J Med Chem. 2011;54(23):8188–94.
Njogu PM, Guantai EM, Pavadai E, Chibale K. Computer-Aided Drug Discovery Approaches against the Tropical Infectious Diseases Malaria, Tuberculosis, Trypanosomiasis, and Leishmaniasis. ACS Infect Disease. 2016;2:8–31.
McCall LI, El Aroussi A, Choi JY, Vieira DF, De Muylder G, Johnston JB, et al. Targeting Ergosterol Biosynthesis in Leishmania donovani: Essentiality of Sterol 14alpha-demethylase. PLoS Negl Trop Dis. 2015;9(3):e0003588.
Sanchez LM, Knudsen GM, Helbig C, De Muylder G, Mascuch SM, Mackey ZB, et al. Examination of the mode of action of the almiramide family of natural products against the kinetoplastid parasite Trypanosoma brucei. J Nat Prod. 2013;76(4):630–41.
Ben Khalaf N, De Muylder G, Louzir H, McKerrow J, Chenik M. Leishmania major protein disulfide isomerase as a drug target: enzymatic and functional characterization. Parasitol Res. 2012;110(5):1911–7.
De Muylder G, Ang KK, Chen S, Arkin MR, Engel JC, McKerrow JH. A screen against Leishmania intracellular amastigotes: comparison to a promastigote screen and identification of a host cell-specific hit. PLoS Negl Trop Dis. 2011;5(7):e1253.
Swenerton RK, Zhang S, Sajid M, Medzihradszky KF, Craik CS, Kelly BL, et al. The oligopeptidase B of Leishmania regulates parasite enolase and immune evasion. J Biol Chem. 2011;286(1):429–40.
Chen CK, Leung SS, Guilbert C, Jacobson MP, McKerrow JH, Podust LM. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl Trop Dis. 2010;4(4):e651.
Zhang S, Kim CC, Batra S, McKerrow JH, Loke P. Delineation of diverse macrophage activation programs in response to intracellular parasites and cytokines. PLoS Negl Trop Dis. 2010;4(3):e648.
McKerrow JH, Rosenthal PJ, Swenerton R, Doyle P. Development of protease inhibitors for protozoan infections. Curr Opin Infect Dis. 2008;21(6):668–72.
Fricker SP, Mosi RM, Cameron BR, Baird I, Zhu Y, Anastassov V, et al. Metal compounds for the treatment of parasitic diseases. J Inorg Biochem. 2008;102(10):1839–45.
McKerrow JH, Caffrey C, Kelly B, Loke P, Sajid M. Proteases in parasitic diseases. Annu Rev Pathol. 2006;1:497–536.
Mahmoudzadeh-Niknam H, McKerrow JH. Leishmania tropica: cysteine proteases are essential for growth and pathogenicity. Exp Parasitol. 2004;106(3-4):158–63.
Selzer PM, Pingel S, Hsieh I, Ugele B, Chan VJ, Engel JC, et al. Cysteine protease inhibitors as chemotherapy: lessons from a parasite target. Proc Natl Acad Sci U S A. 1999;96(20):11015–22.
Scheidt KA, Roush WR, McKerrow JH, Selzer PM, Hansell E, Rosenthal PJ. Structure-based design, synthesis and evaluation of conformationally constrained cysteine protease inhibitors. Bioorg Med Chem. 1998;6(12):2477–94.
McKerrow JH, Sun E, Rosenthal PJ, Bouvier J. The proteases and pathogenicity of parasitic protozoa. Annu Rev Microbiol. 1993;47:821–53.
Nuhs A, De Rycker M, Manthri S, Comer E, Scherer CA, Schreiber SL, et al. Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Negl Trop Dis. 2015;9(9):e0004094.
Pena I, Pilar Manzano M, Cantizani J, Kessler A, Alonso-Padilla J, Bardera AI, et al. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: an open resource. Sci Rep. 2015;5:8771.
Kaiser M, Maes L, Tadoori LP, Spangenberg T, Ioset JR. Repurposing of the Open Access Malaria Box for Kinetoplastid Diseases Identifies Novel Active Scaffolds against Trypanosomatids. J Biomol Screen. 2015;20(5):634–45.
Hotez PJ. Preface. In: Caffrey CR, editor. Parasitic Helminths Targets, Screens, Drugs and Vaccines. Weinheim: Wiley-Blackwell; 2012. p. XI
Ekpo UF, Oluwole AS, Abe EM, Etta HE, Olamiju F, Mafiana CF. Schistosomiasis in infants and pre-school-aged children in sub-Saharan Africa: implication for control. Parasitology. 2012;139(7):835–41.
Colley DG, Bustinduy AL, Secor WE, King CH. Human schistosomiasis. Lancet. 2014;383(9936):2253–64.
Naing C, Whittaker MA, Nyunt-Wai V, Reid SA, Wong SF, Mak JW, et al. Malaria and soil-transmitted intestinal helminth co-infection and its effect on anemia: a meta-analysis. Trans R Soc Trop Med Hyg. 2013;107(11):672–83.
Kobylinski KC, Alout H, Foy BD, Clements A, Adisakwattana P, Swierczewski BE, et al. Rationale for the Coadministration of Albendazole and Ivermectin to Humans for Malaria Parasite Transmission Control. Am J Trop Med Hyg. 2014.
Caffrey CR. Parasitic Helminths: Targets, Screens, Drugs and Vaccines. Wiley-Blackwell: Weinheim; 2012.
Feldmeier H, Krantz I, Poggensee G. Female genital schistosomiasis as a risk-factor for the transmission of HIV. Int J STD AIDS. 1994;5(5):368–72.
Kjetland EF, Ndhlovu PD, Gomo E, Mduluza T, Midzi N, Gwanzura L, et al. Association between genital schistosomiasis and HIV in rural Zimbabwean women. AIDS. 2006;20(4):593–600.
Christinet V, Lazdins-Helds JK, Stothard JR, Reinhard-Rupp J. Female genital schistosomiasis (FGS): from case reports to a call for concerted action against this neglected gynaecological disease. Int J Parasitol. 2016;46(7):395–404.
Keiser J, Utzinger J. The drugs we have and the drugs we need against major helminth infections. Adv Parasitol. 2010;73:197–230.
Gabrielli AF, Montresor A, Chitsulo L, Engels D, Savioli L. Preventive chemotherapy in human helminthiasis: theoretical and operational aspects. Trans R Soc Trop Med Hyg. 2011;105(12):683–93.
van den Enden E. Pharmacotherapy of helminth infection. Expert Opin Pharmacother. 2009;10(3):435–51.
Caffrey CR, editor. Parasitic Helminths: Targets, Screens, Drugs and Vaccines. Weinheim: Wiley-Blackwell; 2012.
Caffrey CR. Schistosomiasis and its treatment. Future Med Chem. 2015;7(6):675–6.
Panic G, Flores D, Ingram-Sieber K, Keiser J. Fluorescence/luminescence-based markers for the assessment of Schistosoma mansoni schistosomula drug assays. Parasit Vectors. 2015;8:624.
Abdulla MH, Ruelas DS, Wolff B, Snedecor J, Lim KC, Xu F, et al. Drug discovery for schistosomiasis: hit and lead compounds identified in a library of known drugs by medium-throughput phenotypic screening. PLoS Negl Trop Dis. 2009;3(7):e478.
Singh R, Pittas, M., Heskia, I., Xu, F., McKerrow, J. H., Caffrey, C. R. Automated image-based phenotypic screening for high-throughput drug discovery. In.IEEE Symposium on Computer-Based Medical Systems Albuquerque, NM; 2009. p. 1-8.
Lee H, Moody-Davis A, Saha U, Suzuki BM, Asarnow D, Chen S, et al. Quantification and clustering of phenotypic screening data using time-series analysis for chemotherapy of schistosomiasis. BMC Genomics. 2012;13(Suppl 1):S4.
Asarnow D, Rojo-Arreola L, Suzuki BM, Caffrey CR, Singh R. The QDREC web server: determining dose-response characteristics of complex macroparasites in phenotypic drug screens. Bioinformatics. 2014.
Paveley RA, Mansour NR, Hallyburton I, Bleicher LS, Benn AE, Mikic I, et al. Whole organism high-content screening by label-free, image-based Bayesian classification for parasitic diseases. PLoS Negl Trop Dis. 2012;6(7):e1762.
Caffrey CR. Chemotherapy of schistosomiasis: present and future. Curr Opin Chem Biol. 2007;11(4):433–9.
Caffrey CR, Secor WE. Schistosomiasis: from drug deployment to drug development. Curr Opin Infect Dis. 2011;24(5):410–7.
Nwaka S, Ramirez B, Brun R, Maes L, Douglas F, Ridley R. Advancing drug innovation for neglected diseases-criteria for lead progression. PLoS Negl Trop Dis. 2009;3(8):e440.
Andrews P, Thomas H, Pohlke R, Seubert J. Praziquantel. Med Res Rev. 1983;3(2):147–200.
Sabah AA, Fletcher C, Webbe G, Doenhoff MJ. Schistosoma mansoni: chemotherapy of infections of different ages. Exp Parasitol. 1986;61(3):294–303.
Xiao SH, Yue WJ, Yang YQ, You JQ. Susceptibility of Schistosoma japonicum to different developmental stages to praziquantel. Chin Med J. 1987;100(9):759–68.
Ingram-Sieber K, Cowan N, Panic G, Vargas M, Mansour NR, Bickle QD, et al. Orally active antischistosomal early leads identified from the open access malaria box. PLoS Negl Trop Dis. 2014;8(1):e2610.
Fonseca NC, da Cruz LF, Villela FD, Pereira GA, de Siqueira-Neto JL, Kellar D, et al. Synthesis of a sugar-based thiosemicarbazone series and structure-activity relationship versus the parasite cysteine proteases: rhodesain, cruzain and Schistosoma mansoni cathepsin B1. Antimicrob Agents Chemother. 2015.
Rojo-Arreola L, Long T, Asarnow D, Suzuki BM, Singh R, Caffrey CR. Chemical and genetic validation of the statin drug target to treat the helminth disease, schistosomiasis. PLoS One. 2014;9(1):e87594.
Kannan S, Melesina J, Hauser AT, Chakrabarti A, Heimburg T, Schmidtkunz K, et al. Discovery of inhibitors of Schistosoma mansoni HDAC8 by combining homology modeling, virtual screening, and in vitro validation. J Chem Inf Model. 2014;54(10):3005–19.
Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arner ES, et al. Thioredoxin glutathione reductase from Schistosoma mansoni: an essential parasite enzyme and a key drug target. PLoS Med. 2007;4(6):e206.
Neves BJ, Dantas RF, Senger MR, Melo-Filho CC, Valente WC, de Almeida AC, et al. Discovery of New Anti-Schistosomal Hits by Integration of QSAR-Based Virtual Screening and High Content Screening. J Med Chem. 2016;59(15):7075–88.
Long T, Rojo-Arreola L, Shi D, El-Sakkary N, Jarnagin K, Rock F, et al. Phenotypic, chemical and functional characterization of cyclic nucleotide phosphodiesterase 4 (PDE4) as a potential anthelmintic drug target. PLoS Negl Trop Dis. 2017;11(7):e0005680.
Neves BJ, Braga RC, Bezerra JC, Cravo PV, Andrade CH. In silico repositioning-chemogenomics strategy identifies new drugs with potential activity against multiple life stages of Schistosoma mansoni. PLoS Negl Trop Dis. 2015;9(1):e3435.
Neves BJ, Dantas RF, Senger MB, Valente WCG, Rezende-Neto JD, Chaves WT, et al. AC. The antidepressant drug paroxetine as a new lead candidate in schistosome drug discovery. ChemMedChem. 2016;7:1176–82.
Burns AR, Luciani GM, Musso G, Bagg R, Yeo M, Zhang Y, et al. Caenorhabditis elegans is a useful model for anthelmintic discovery. Nat Commun. 2015;6:7485.
Mathew MD, Mathew ND, Miller A, Simpson M, Au V, Garland S, et al. Using C. elegans Forward and Reverse Genetics to Identify New Compounds with Anthelmintic Activity. PLoS Negl Trop Dis. 2016;10(10):e0005058.
Crowther GJ, Booker ML, He M, Li T, Raverdy S, Novelli JF, et al. Cofactor-independent phosphoglycerate mutase from nematodes has limited druggability, as revealed by two high-throughput screens. PLoS Negl Trop Dis. 2014;8(1):e2628.
Bilsland E, Bean DM, Devaney E, Oliver SG. Yeast-Based High-Throughput Screens to Identify Novel Compounds Active against Brugia malayi. PLoS Negl Trop Dis. 2016;10(1):e0004401.
Stadelmann B, Rufener R, Aeschbacher D, Spiliotis M, Gottstein B, Hemphill A. Screening of the Open Source Malaria Box Reveals an Early Lead Compound for the Treatment of Alveolar Echinococcosis. PLoS Negl Trop Dis. 2016;10(3):e0004535.
Pothineni VR, Wagh D, Babar MM, Inayathullah M, Solow-Cordero D, Kim KM, et al. Identification of new drug candidates against Borrelia burgdorferi using high-throughput screening. Drug Des Devel Ther. 2016;10:1307–22.
Boyom FF, Fokou PV, Tchokouaha LR, Spangenberg T, Mfopa AN, Kouipou RM, et al. Repurposing the open access malaria box to discover potent inhibitors of Toxoplasma gondii and Entamoeba histolytica. Antimicrob Agents Chemother. 2014;58(10):5848–54.
Witola WH, Mui E, Hargrave A, Liu S, Hypolite M, Montpetit A, et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun. 2011;79(2):756–66.
Cruz DJ, Koishi AC, Taniguchi JB, Li X, Milan Bonotto R, No JH, et al. High content screening of a kinase-focused library reveals compounds broadly-active against dengue viruses. PLoS Negl Trop Dis. 2013;7(2):e2073.
WHO. Dengue and severe dengue. Available from: http://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue.
Lim SP, Wang QY, Noble CG, Chen YL, Dong H, Zou B, et al. Ten years of dengue drug discovery: progress and prospects. Antivir Res. 2013;100(2):500–19.
Yang CC, Hu HS, Wu RH, Wu SH, Lee SJ, Jiaang WT, et al. A novel dengue virus inhibitor, BP13944, discovered by high-throughput screening with dengue virus replicon cells selects for resistance in the viral NS2B/NS3 protease. Antimicrob Agents Chemother. 2014;58(1):110–9.
Cheung YY, Chen KC, Chen H, Seng EK, Chu JJ. Antiviral activity of lanatoside C against dengue virus infection. Antivir Res. 2014;111:93–9.
Basavannacharya C, Vasudevan SG. Suramin inhibits helicase activity of NS3 protein of dengue virus in a fluorescence-based high throughput assay format. Biochem Biophys Res Commun. 2014;453(3):539–44.
Chu JJ, Yang PL. c-Src protein kinase inhibitors block assembly and maturation of dengue virus. Proc Natl Acad Sci U S A. 2007;104(9):3520–5.
Faye O, Freire CC, Iamarino A, Faye O, de Oliveira JV, Diallo M, et al. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl Trop Dis. 2014;8(1):e2636.
Coffee M. Understanding the Zika Outreak and Why It's Rapidly Spreading. Available from: http://infectiousdiseases.about.com/od/travelershealth/fl/Zika.htm.
Oehler E, Watrin L, Larre P, Leparc-Goffart I, Lastere S, Valour F, et al. Zika virus infection complicated by Guillain-Barre syndrome--case report, French Polynesia, December 2013. Euro Surveill. 2014;19(9).
Smith DW, Mackenzie J. Zika virus and Guillain-Barre syndrome: another viral cause to add to the list. Lancet. 2016;387(10027):1486–8.
Faria NR, Azevedo Rdo S, Kraemer MU, Souza R, Cunha MS, Hill SC, et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science. 2016;352(6283):345–9.
Anon. WHO Director-General summarizes the outcome of the Emergency Committee regarding clusters of microcephaly and Guillain-Barré syndrome. Available from: http://who.int/mediacentre/news/statements/2016/emergency-committee-zika-microcephaly/en/.
Ekins S, Mietchen D, Coffee M, Stratton TP, Freundlich JS, Freitas-Junior L, et al. Open drug discovery for the Zika virus. F1000Res. 2016;5:150.
Shan C, Xie X, Barrett AD, Garcia-Blanco MA, Tesh RB, Vasconcelos PF, et al. Zika Virus: Diagnosis, Therapeutics, and Vaccine. ACS Infect Dis. 2016;2:170–2.
Malone RW, Homan J, Callahan MV, Glasspool-Malone J, Damodaran L, Schneider Ade B, et al. Zika Virus: Medical Countermeasure Development Challenges. PLoS Negl Trop Dis. 2016;10(3):e0004530.
Mottin M, Borba JVVB, Melo-Filho CC, Neves BJ, Muratov E., Torres PHM, Braga RC, Perryman A, Ekins S, Andrade CH. Computational Drug Discovery for the Zika Virus. Brazilian Journal of Pharmaceutical Sciences. 2018;In Press.
Ekins S, Perryman AL, Horta AC. OpenZika: An IBM World Community Grid Project to Accelerate Zika Virus Drug Discovery. PLoS Negl Trop Dis. 2016;10(10):e0005023.
Mottin M, Borba J, Braga RC, Torres PHM, Martini MC, Proenca-Modena JL, Judice CC, Costa FTM, Ekins S, Perryman AL, Horta Andrade C. The A-Z of Zika drug discovery. Drug Discov Today. 2018.
Xu M, Lee EM, Wen Z, Cheng Y, Huang WK, Qian X, et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat Med. 2016;22(10):1101–7.
Retallack H, Di Lullo E, Arias C, Knopp KA, Laurie MT, Sandoval-Espinosa C, et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc Natl Acad Sci U S A. 2016;113(50):14408–13.
Ekins S, Southan C, Coffee M. Finding small molecules for the 'next Ebola'. F1000Res. 2015;4:58.
Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, et al. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med. 2013;5(190)):190ra179–9.
Towner JS, Paragas J, Dover JE, Gupta M, Goldsmith CS, Huggins JW, et al. Generation of eGFP expressing recombinant Zaire ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology. 2005;332(1):20–7.
Cheng H, Lear-Rooney CM, Johansen L, Varhegyi E, Chen ZW, Olinger GG, et al. Inhibition of Ebola and Marburg Virus Entry by G Protein-Coupled Receptor Antagonists. J Virol. 2015;89(19):9932–8.
Ekins S, Freundlich JS, Clark AM, Anantpadma M, Davey RA, Madrid P. Machine learning models identify molecules active against the Ebola virus in vitro. F1000Res. 2016;4:1091.
Ekins S, Lingerfelt MA, Comer JE, Freiberg AN, Mirsalis JC, O’Loughlin K, Harutyunyan A, McFarlane C, Green CE, Madrid PB. Efficacy of Tilorone Dihydrochloride against Ebola Virus Infection. Antimicrob Agents Chemother. 2017;In Press.
Mirza MU, Ikram N. Integrated Computational Approach for Virtual Hit Identification against Ebola Viral Proteins VP35 and VP40. Int J Mol Sci. 2016;17(11).
Chopra G, Kaushik S, Elkin PL, Samudrala R. Combating Ebola with Repurposed Therapeutics Using the CANDO Platform. Molecules. 2016;21(12).
Mudhasani R, Kota KP, Retterer C, Tran JP, Whitehouse CA, Bavari S. High content image-based screening of a protease inhibitor library reveals compounds broadly active against Rift Valley fever virus and other highly pathogenic RNA viruses. PLoS Negl Trop Dis. 2014;8(8):e3095.
Cruz DJ, Bonotto RM, Gomes RG, da Silva CT, Taniguchi JB, No JH, et al. Identification of novel compounds inhibiting chikungunya virus-induced cell death by high throughput screening of a kinase inhibitor library. PLoS Negl Trop Dis. 2013;7(10):e2471.
Karlas A, Berre S, Couderc T, Varjak M, Braun P, Meyer M, et al. A human genome-wide loss-of-function screen identifies effective chikungunya antiviral drugs. Nat Commun. 2016;7:11320.
Dyall J, Coleman CM, Hart BJ, Venkataraman T, Holbrook MR, Kindrachuk J, et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother. 2014;58(8):4885–93.
Oprea TI, Allu TK, Fara DC, Rad RF, Ostopovici L, Bologa CG. Lead-like, drug-like or "Pub-like": how different are they? J Comput Aided Mol Des. 2007;21(1-3):113–9.
Lane T, Russo DP, Zorn KM, Clark AM, Korotcov A, Tkachenko V, Reynolds RC, Perryman AL, Freundlich JS, Ekins S. Comparing and Validating Machine Learning Models for Mycobacterium tuberculosis Drug Discovery. Mol Pharm. 2018.
Russo DP, Zorn KM, Clark AM, Zhu H, Ekins S. Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol Pharmaceutics. 2018.
Sandoval PJ, Zorn KM, Clark AM, Ekins S, Wright SH. Assessment of Substrate Dependent Ligand Interactions at the Organic Cation Transporter OCT2 Using Six Model Substrates. Mol Pharmacol. 2018.
Ekins S, Freundlich J, Clark A, Anantpadma M, Davey R, Madrid P. Machine learning models identify molecules active against Ebola virus in vitro. F1000Res. 2015;4:1091.
Guiguemde WA, Shelat AA, Garcia-Bustos JF, Diagana TT, Gamo FJ, Guy RK. Global phenotypic screening for antimalarials. Chem Biol. 2012;19(1):116–29.
Tarral A, Blesson S, Mordt OV, Torreele E, Sassella D, Bray MA, et al. Determination of an optimal dosing regimen for fexinidazole, a novel oral drug for the treatment of human African trypanosomiasis: first-in-human studies. Clin Pharmacokinet. 2014;53(6):565–80.
Raether W, Seidenath H. The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, trichomonads and Entamoeba histolytica. Ann Trop Med Parasitol. 1983;77(1):13–26.
Bahia MT, de Andrade IM, Martins TA, do Nascimento AF, Diniz Lde F, Caldas IS, et al. Fexinidazole: a potential new drug candidate for Chagas disease. PLoS Negl Trop Dis. 2012;6(11):e1870.
Acknowledgments and Disclosures
This work was supported by Award Numbers 1R43GM122196-01 and R44GM122196-02A1 “Centralized assay datasets for modelling support of small drug discovery organizations” from NIH/ NIGMS. 1UH2TR002084-01 “Repurposing pyronaridine as a treatment for Chagas disease” from NIH/ NCATS 1R21TR001718-01 “Repurposing pyronaridine as a treatment for the Ebola virus” from NIH / NCATS. MM thanks the support of the Brazilian CNPq/FAPEG DCR (grant 300508/2017-4). We are very grateful for our many collaborators and colleagues who contributed to the various efforts referenced herein. HWH is a Managing Member of KAL Research Initiatives, LLC. KMZ is an employee and SE is an employee and owner of Collaborations Pharmaceuticals, Inc.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hernandez, H.W., Soeung, M., Zorn, K.M. et al. High Throughput and Computational Repurposing for Neglected Diseases. Pharm Res 36, 27 (2019). https://doi.org/10.1007/s11095-018-2558-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11095-018-2558-3