
Two series of new aromatic thiosemicarbazone derivatives were synthesized by condensation of N-(4-cyanophenyl)hydrazine carbothioamide (I) and N-(4-methylsulfanylphenyl)hydrazine carbothioamide (II) with appropriate aromatic aldehydes in order to investigate their antiviral and cytostatic potency. The chemical structures of all compounds were fully characterized by elemental analysis and spectroscopic techniques. The results of the bioassays indicated that compounds Id, Ie, If and IIf proved inhibitory against influenza virus A (EC50 = 13 – 27 μg/mL for strain H1N1 and 9.3 – 18 μg/mL for strain H3N2). Compounds Ig and IIg were the most cytostatic compounds with inhibition of HeLa cell proliferation at an IC50 = 0.3 μg/mL for Ig and 1.9 μg/mL for IIg. Especially, compound Ig showed the highest cytostatic activity with IC50 of 0.30, 0.70 and 2.50 μg/mL against HeLa, CEM and L1210 cell lines, respectively. This inhibition range was within the same order of magnitude as that for cisplatin. Furthermore, molecular modeling was carried out to examine the cytostatic activity and determine the best pharmacophore model as a guide for the design and development of potential prodrugs in future studies.
Similar content being viewed by others

1. Introduction
Thiosemicarbazones are important class of synthetic products and potential biologically active compounds. Domagk, et al. [1] first reported that thiosemicarbazone pharmacophore had antituberculosis activity. Then, antiviral activity of benzaldehyde thiosemicarbazone derivatives against vaccina virus in mice was found by Hamre, et al. [2]. The antiviral activity of isatin-β-thiosemicarbazone (IBT) and N-methyl-isatin-β-thiosemicarbazone (commercially known as Methisazone or Marboran) was widely investigated against orthopoxviruses in the 1960s [3]. After these discoveries, thiosemicarbazones draw considerable interest due to their application in the pharmaceutical chemistry and proved to be chemotherapeutic agents potentially useful for inhibiting cancer cells [4]. For example, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (commercially known as Triapine) inhibited the biosynthesis of DNA in murine leukemia L1210 cells by blocking the activity of ribonucleotide reductase [5].
In recent years, it has been commonly accepted that agents containing more than one pharmacophore can have superior efficacy as compared to single-pharmacophore drugs [6]. Pharmacophore hybridization is a method of rational drug design, and a single molecule containing different modes of action can be beneficial for the treatment of diseases. As an important pharmacophore, bis-alkylating nitrogen mustard such as chlormethine, melphalan, chlorambucil and many more have anticancer activities against hematologic tumors, myeloma, ovarian cancer and solid tumors [7]. Alkylating agents are electrophilic entities that interact with nucleophilic moieties of DNA resulting in the covalent transfer of an alkyl group. Another pharmacophore para-substituted aryl nitrile, which is bioisostere of a ketone group, has a variety of anticancer activities, e.g., against hormonally-responsive breast cancer (letrozole), breast cancer (neratinib), prostate cancer (bicalutamide), pancreatic cancer, non-small cell lung cancer, head and neck cancer (L-778,123, phase I), and chronic myelogenous leukemia (bosutinib, SKI-606, phase III) [8].
Previously, we reported in vitro cytostatic and broad-spectrum antiviral activity of the thiosemicarbazones derived from 5-substitutedthiophene-2-carboxaldehydes and their platinum(II) and palladium(II) complexes [9]. Encouraged by these results and aimed at developing effective anticancer agents, we designed two series of thiosemicarbazones by the pharmacophore hybridization method using two or more different pharmacophores in view of prospecting their cytostatic and antiviral activity (Fig. 1). In order to gain detailed information about this issue and to develop effective anticancer agents for future studies, we have performed virtual screening of the aforementioned compounds by using Gaussian09 and Discovery Studio 3.5 software [10, 11].

Design of compound Ig.
The intermediates N-(4-cyanophenyl)hydrazinecarbothioamide (I) and N-(4-methylthiophenyl)hydrazinecarbothioamide (II) were prepared from 4-cyanophenyl isothiocyanate and 4-methylthiophenyl isothiocyanate by reaction with hydrazine monohydrate, respectively [12]. N-(4-cyanophenyl) hydrazinecarbothioamide (I) was reported previously [13] and N-(4-methylthiophenyl)hydrazinecarbothioamide (II) was commercially available. The novel thiosemicarbazones (Ia – Ig and IIa – IIg) as well as the known analog of Ib [14] were synthesized in high yield (71 – 92%) from the corresponding 4-substitutedbenzaldehydes by treatment with thiosemicarbazides in methanol, according to the general procedure of a previously described method [15]. The synthetic route of target compounds Ia – Ig and IIa – IIg is shown in Scheme 1.

Synthetic pathway of thiosemicarbazones Ia – Ig and IIa – IIg. Reagents and conditions: (a) NH2NH2.H2O (98%), Et2O, rt, 24 h; (b) appropriate aldeyhde in MeOH, reflux.
The chemical structures of new compounds were throughly eludicated by elemental analysis and spectral data (UV-Vis, IR, 1H NMR, 13C NMR, and ESI-MS). Assignments of selected diagnostic IR bands provided significant indication for the formation of the thiosemicarbazone derivaderivatives. All thiosemicarbazone derivatives (Ia-g and IIa-g) exhibited two intense bands in the region of 3396 – 3266 cm-1 and 3162 – 3120 cm-1 due to ν(N-H) stretching. The strong band at 839 – 818 cm-1 was present due to ν(C=S) stretching. It was suggested that thiosemicarbazones in the solid phase remain in the thione form. In addition, the characteristic azomethine stretching vibrations ν(C=N) at 1587 – 1543 cm-1 were observed, which also confirmed the formation of thiosemicarbazones [16]. Results of the 1H NMR integrations and signal multiplicities were in line with the proposed structures and other spectral data. The 1H NMR spectra of Ia – Ig and IIa – IIg showed a singlet peak attributable to the =N-NH proton in arange of δ = 12.60 – 11.62 ppm as well as a singlet peak attributable to the PhNH proton in the range δ = 10.80 – 9.92 ppm. The signal of the azomethine proton (HC=N) appeared as a singlet at δ = 8.63 – 8.04 ppm [16]. All aromatic protons were observed with the expected chemical shift (δ = 8.48 – 6.80 ppm) and coupling constant in the NMR spectra of all thiosemicarbazone derivatives. The 13C NMR spectra of the thiosemicarbazones exhibited two important signals at δ = 176.92 – 174.23 and at δ = 143.89 – 142.57 ppm assigned to thioamide (C=S) and imine (C=N) carbon atoms, respectively. The signals at δ = 164.01 – 112.06 ppm in the spectra were assumed to be due to the aromatic carbons. The MS spectra of all thiosemicarbazones were in line with the proposed structures.
2. Experimental Chemical Part
The progress of all reactions were monitored by thin layer chromatography (TLC). TLC was performed on silica gel plates (Merck Silica Gel 60, F254, 0.2 mm) with visualization by exposure to iodine vapor and UV light using EtOAc/hexane (v/v 1:1 and 1:3) as solvent system. Melting points were determined on a EZ-Melt MPA120 Automated Melting Point apparatus and were uncorrected. The IR spectra were recorded on a Perkin Elmer 100 FT-IR spectrometer with universal ATR sampling accessory. The 1H NMR and 13C NMR spectra were obtained at room temperature with a Bruker Avance-DPX-400 NMR spectrometer in DMSO-d6 using TMS as the internal standard. The mass spectra were obtained using an LC/MS Agilent 1100 MSD series spectrometer in the electrospray mode. Elemental (CHNS) analyses were performed using a VarioMICRO elemental analyzer.
2.1. Synthesis of Thiosemicarbazides I and II
General procedure. A solution of hydrazine monohydrate (7.50 mmol) in diethylether (20 mL) was added dropwise to a solution of 4-substitutedphenyl isothiocyanate (6.25 mmol) in diethyl ether (20 mL) at room temperature with vigorous stirring. The mixture was allowed to stand overnight. The precipitated solid was filtered and washed two times with water. The crude product was recrystallized from methanol.
N-(4-cyanophenyl)hydrazinecarbothioamide (I). Light yellow solid (methanol). Yield: 1.14 g (95%); m.p.: 185 – 186°C. Anal. calc. for C8H8N4S: C, 49.98; H, 4.19; N, 29.14; S, 16.68%. Found: C, 49.86; H, 4.29; N, 29.16; S, 16.84%. UV (DMSO, λmax, nm): 325; 293; 236. IR (νmax, cm-1): 3338, 3196 (N-H); 2229 (C≡N); 1208 (C-N); 1083 (N-N); 831 (C=S). 1H NMR (DMSO-d6; δ, ppm): 9.53 (s, 1H, NHNH2); 9.40 (s, 1H, PhNH); 8.10 (d, 2H, ArH, J = 8.23 Hz, orto protons to -CN group); 7.76 (d, 2H, ArH, J = 8.26 Hz, meta protons to -CN group); 4.99 (brs, 2H, NH2).
N-(4-methylsulfanylphenyl)hydrazinecarbothioamide (II). White solid (methanol). Yield: 1.17 g (88%); m.p.: 181 – 182°C. Anal. calc. for C8H11N3S2: C, 45.04; H, 5.20; N, 19.70; S, 30.06%. Found: C, 45.24; H, 5.43; N, 19.63; S, 30.07%. UV (DMSO, λmax, nm): 330; 293; 237. IR (νmax, cm-1): 3346, 3274, 3175 (N-H); 1203 (C-N); 1064 (N-N); 828 (C=S). 1H NMR (DMSO-d6; δ, ppm): 9.60 (s, 1H, NHNH2); 9.12 (s, 1H, PhNH); 7.59 (d, 2H, ArH, J = 8.57 Hz, meta protons to -SCH3 group); 7.21 (d, 2H, ArH, J = 8.61 Hz, orto protons to -SCH3 group); 4.80 (brs, 2H, NH2); 2.46 (s, 3H, SCH3).
2.2. Synthesis of Thiosemicarbazones Ia – Ig and IIa – IIg
General procedure: To a hot solution of thiosemicarbazide (1.04 mmol) in methanol (25 mL) was added dropwise a solution of the appropriate aldehyde (1.04 mmol) in methanol (10 mL) with continuous stirring. After the addition of a catalytic amount of glacial acetic acid, the reaction mixture was refluxed for 6 – 24 h. The progress of the reaction was monitored by TLC. The reaction mixture was cooled and the precipitate was filtered. The crude product was washed with cold diethylether or ethanol and recrystallized from appropriate solvent. Table 1 presents data of mass spectroscopy and elemental analysis for aromatic thiosemicarbazones Ia – Ig and Iýa – IIg.
2-Benzylidene-N-(4-cyanophenyl)hydrazinecarbothioamide (Ia). Yellow solid (acetone). Yield: 0.26 g (90%); m.p.: 193 – 194°C. IR (νmax, cm-1): 3275, 3140 (N-H); 2220 (C≡N); 1580 (C=N); 1200 (C-N); 1063 (N-N); 834 (C=S). 1H NMR (DMSO-d6; δ, ppm): 12.10 (s, 1H, CSNH); 10.33 (s, 1H, PhNH); 8.22 (s, 1H, HC=N); 7.97 (d, 2H, ArH, J = 8.62 Hz, orto protons to -CN group); 7.91 (dd, 2H, ArH, J1 = 3.58 Hz, J2 = 2.40 Hz, orto protons at phenyl ring); 7.83 (d, 2H, ArH, J = 8.62 Hz, meta protons to -CN group); 7.46 (t, 3H, ArH, J1 = 3.42 Hz, J2 = 2.93 Hz, para and meta protons at phenyl ring). 13C NMR (DMSO-d6; δ, ppm): 176.49 (C=S), 143.78 (C=N), 141.99, 133.83, 132.34, 131.75, 130.16, 129.13, 128.55, 118.67 (ArC), 108.62 (C≡N).
2-(4-Hydroxybenzylidene)-N-(4-cyanophenyl)hydrazinecarbothioamide (Ib). Yellow solid (acetone). Yield: 0.22 g (71%); m.p.: 224 – 225°C. IR (νmax, cm-1): 3396, 3153 (O-H and N-H); 2229 (C≡N); 1579 (C=N); 1207 (C-N); 1060 (N-N); 833 (C=S). 1H NMR (DMSO-d6; δ, ppm): 11.93 (s, 1H, CSNH); 10.20 (s, 1H, PhNH); 9.98 (s, 1H, OH); 8.10 (s, 1H, HC=N); 7.98 (d, 2H, ArH, J = 8.69 Hz, orto protons to -CN group); 7.82 (d, 2H, ArH, J = 8.67 Hz, meta protons to -CN group); 7.74 (d, 2H, ArH, J = 8.64 Hz, meta protons to -OH group); 6.82 (d, 2H, ArH, J = 8.64 Hz orto protons to -OH group). 13C NMR (DMSO-d6; δ, ppm): 175.56 (C=S), 154.42 (ArC, ipso carbon to -OH group), 143.48 (C=N), 141.98, 132.64, 131.73, 130.21, 127.47, 119.44, 116.72 (ArC), 107.52 (C≡N).
2-(4-Methoxybenzylidene)-N-(4-cyanophenyl)hydrazinecarbothioamide (Ic). Beige solid (ethanol). Yield: 0.29 g (92%); m.p.: 217 – 218°C. IR (νmax, cm-1): 3273, 3158 (N-H); 2227 (C≡N); 1543 (C=N); 1201 (C-N); 1022 (N-N); 835 (C=S). 1H NMR (DMSO-d6; δ, ppm): 12.03 (s, 1H, CSNH); 10.25 (s, 1H, PhNH); 8.15 (s, 1H, HC=N); 7.97 (d, 2H, ArH, J = 8.73 Hz, orto protons to -CN group); 7.85 (d, 2H, ArH, J = 8.83 Hz, meta protons to -OCH3 group); 7.82 (d, 2H, ArH, J = 8.70 Hz, meta protons to -CN group); 7.01 (d, 2H, ArH, J = 8.81 Hz, orto protons to -OCH3 group); 3.81 (s, 3H, OCH3). 13C NMR (DMSO-d6; δ, ppm): 176.38 (C=S), 158.32 (ArC, ipso carbon to -OCH3 group), 143.76 (C=N), 142.58, 140.35, 133.09, 129.17, 126.76, 124.86, 119.49 (ArC), 106.98 (C≡N), 56.12 (OCH3).
2-[(4-Trifluoromethoxy)benzylidene]-N-(4-cyanophenyl)hydrazinecarbothioamide (Id). White solid (acetone). Yield: 0.28 g (72%); m.p.: 219 – 220°C. IR (νmax, cm-1): 3291, 3149 (N-H); 2223 (C≡N); 1549 (C=N); 1201 (C-N); 1079 (N-N); 839 (C=S). 1H NMR (DMSO-d6; δ, ppm): 12.60 (s, 1H, CSNH); 10.80 (s, 1H, PhNH); 8.63 (s, 1H, HC=N); 8.48 (d, 2H, ArH, J = 8.27 Hz, meta protons to -OCF3 group); 8.37 (d, 2H, ArH, J = 8.65 Hz, orto protons to –CN group); 8.27 (d, 2H, ArH, J = 8.66 Hz, meta protons to –CN group); 7.86 (d, 2H, ArH, J = 8.22 Hz, orto protons to -OCF3 group). 13C NMR (DMSO-d6; δ, ppm): 176.24 (C=S), 146.45 (ArC, ipso carbon to OCF3), 142.76 (C=N), 140.37, 139.63, 131.85, 128.79 (ArC), 126.41 (OCF3), 124.19, 123.07, 115.54 (ArC), 107.05 (C≡N).
2-(4-Fluorobenzylidene)-N-(4-cyanophenyl)hydrazinecarbothioamide (Ie). White solid (acetone). Yield: 0.23 g (73%); m.p.: 206 – 207°C. IR (νmax, cm-1): 3284, 3146 (N-H); 2226 (C≡N); 1581 (C=N); 1201 (C-N); 1076 (N-N); 827 (C=S). 1H NMR (DMSO-d6; δ, ppm): 12.51 (s, 1H, CSNH); 10.73 (s, 1H, PhNH); 8.61 (s, 1H, HC=N); 8.42 (dd, 2H, ArH, J = 8.79 Hz, meta protons to -F atom); 8.37 (d, 2H, ArH, J = 8.74 Hz, orto protons to -CN group); 8.26 (d, 2H, ArH, J = 8.73 Hz, meta protons to -CN group); 7.72 (t, 2H, ArH, J = 8.86 Hz, orto protons to -F atom). 13C NMR (DMSO-d6; δ, ppm): 176.71 (C=S), 163.99 (ArC, ipso carbon to -F atom), 142.77 (C=N), 139.78, 134.14, 132.42, 130.23, 126.47, 118.53, 116.77 (ArC), 106.96 (C≡N).
2-(4-Nitrobenzylidene)-N-(4-cyanophenyl)hydrazinecarbothioamide (If). Dark yellow solid (ethanol). Yield: 0.27 g (81%); m.p.: 256 – 257°C. IR (νmax, cm-1): 3280, 3140 (N-H); 2224 (C≡N); 1579 (C=N); 1499 (NO2, asymmetric); 1337 (NO2, symmetric); 1200 (C-N); 1088 (N-N); 839 (C=S). 1H NMR (DMSO-d6; δ, ppm): 12.35 (s, 1H, CSNH); 10.51 (s, 1H, PhNH); 8.31 (d, 2H, ArH, J = 8.86 Hz, orto protons to -NO2 group); 8.28 (s, 1H, HC=N); 8.21 (d, 2H, ArH, J = 8.87 Hz, meta protons to -NO2 group); 7.95 (d, 2H, ArH, J = 8.62 Hz, orto protons to -CN group); 7.82 (d, 2H, ArH, J = 8.65 Hz, meta protons to -CN group). 13C NMR (DMSO-d6; δ, ppm): 176.39 (C=S); 148.23 (ArC, ipso carbon to -NO2 group); 143.64 (C=N); 141.62, 140.55, 132.71, 128.97, 125.88, 124.17, 119.27 (ArC), 107.53 (C≡N).
2-{4-[Bis(2-chloroethyl)amino]benzylidene}-N-(4-cyanophenyl)hydrazinecarbo thioamide (Ig). Light brown solid (acetone). Yield: 0.33 g (76%); m.p.: 174 – 175°C. IR (νmax, cm-1): 3290, 3148 (N-H); 2228 (C≡N); 1582 (C=N); 1199 (C-N); 1076 (N-N); 829 (C=S); 719 (C-Cl). 1H NMR (DMSO-d6; δ, ppm): 11.90 (s, 1H, CSNH); 10.18 (s, 1H, PhNH); 8.08 (s, 1H, HC=N); 7.99 (d, 2H, ArH, J = 8.50 Hz, orto protons to -CN group); 7.81 (d, 2H, ArH, J = 8.53 Hz, meta protons to -CN group); 7.73 (d, 2H, ArH, J = 8.70 Hz, meta protons to -N(CH2CH2Cl)2 group); 6.81 (d, 2H, ArH, J = 8.75 Hz, orto protons to -N(CH2CH2Cl)2 group); 3.76 – 3.81 (m, 8H, -N(CH2CH2Cl)2). 13C NMR (DMSO-d6; δ, ppm): 176.49 (C=S), 149.25 (ArC, ipso carbon to -N(CH2CH2Cl)2 group), 143.33 (C=N), 136.64, 134.12, 130.45, 127.68, 125.96, 123.87, 112.65 (ArC), 107.78 (C≡N), 53.18 (N(CH2CH2Cl)2), 42.54 (N(CH2CH2Cl)2).
2-Benzylidene-N-[(4-methylthio)phenyl]hydrazinecarbothioamide (IIa). White solid (acetone). Yield: 0.27 g (85%); m.p.: 211 – 212°C. IR (νmax, cm-1): 3299, 3149 (N-H); 1573 (C=N); 1082 (C-N); 1062 (N-N); 823 (C=S). 1H NMR (DMSO-d6; δ, ppm): 11.84 (s, 1H, CSNH); 10.09 (s, 1H, PhNH); 8.17 (s, 1H, HC=N); 7.91 (dd, 2H, ArH, J1 = 3.80 Hz, J2 =2.05 Hz, orto protons at phenyl ring); 7.53 (d, 2H, ArH, J = 8.58 Hz, meta protons to -SCH3 group); 7.44 (t, 3H, ArH, J1 = 3.47 Hz, J2 =2.27 Hz, para and meta protons at phenyl ring); 7.28 (d, 2H, ArH, J = 8.59 Hz, orto protons to -SCH3 group), 2.49 (s, 3H, SCH3). 13C NMR (DMSO-d6; δ, ppm): 174.23 (C=S), 143.85 (C=N), 135.47, 134.92, 133.76, 131.67, 131.01, 129.28, 128.85, 125.71 (ArC), 15.43 (SCH3).
2-(4-Hydroxybenzylidene)-N-[(4-methylthio)phenyl]hydrazinecarbothioamide (IIb). White solid (methanol). Yield: 0.26 g (81%); m.p.: 202 – 203°C. IR (νmax, cm-1): 3580 (O-H); 3266, 3153 (N-H); 1583 (C=N); 1164 (C-N); 1069 (N-N); 818 (C=S). 1H NMR (DMSO-d6; δ, ppm): 11.65 (s, 1H, CSNH); 9.94 (s, 1H, PhNH); 9.92 (s, 1H, OH); 8.07 (s, 1H, HC=N); 7.73 (d, 2H, ArH, J = 8.59 Hz, meta protons to -OH group); 7.54 (d, 2H, ArH, J = 8.57 Hz, meta protons to -SCH3 group); 7.26 (d, 2H, ArH, J = 8.59 Hz, orto protons to -OH group); 6.81 (d, 2H, ArH, J = 8.57 Hz orto protons to -SCH3 group), 2.49 (s, 3H, SCH3). 13C NMR (DMSO-d6; δ, ppm): 176.92 (C=S), 153.29 (ArC, ipso carbon to –OH group), 142.57 (C=N), 131.27, 129.62, 128.51, 126.13, 125.48, 120.03, 115.68 (ArC), 16.67 (SCH3).
2-(4-Methoxyybenzylidene)-N-[(4-methylthio)phenyl]hydrazinecarbothioamide (IIc). White solid (acetone). Yield: 0.25 g (73%); m.p.: 194 – 195°C. IR (νmax, cm-1): 3309, 3126 (N-H); 1570 (C=N); 1164 (C-N); 1068 (N-N); 823 (C=S). 1H NMR (DMSO-d6; δ, ppm): 11.72 (s, 1H, CSNH); 10.01 (s, 1H, PhNH); 8.11 (s, 1H, HC=N); 7.85 (d, 2H, ArH, J = 8.74 Hz, meta protons to -SCH3 group); 7.53 (d, 2H, ArH, J = 8.57 Hz, meta protons to -OCH3 group); 7.27 (d, 2H, ArH, J = 8.57 Hz, orto protons to -OCH3 group); 6.99 (d, 2H, ArH, J = 8.74 Hz, orto protons to -SCH3 group); 3.81 (s, 3H, OCH3), 2.49 (s, 3H, SCH3). 13C NMR (DMSO-d6; δ, ppm): 175.93 (C=S), 161.25 (ArC, ipso carbon to -OCH3 group), 143.36 (C=N), 136.67, 134.83, 129.66, 126.83, 126.73, 126.10, 114.51 (ArC), 55.70 (OCH3), 15.44 (SCH3).
2-[(4-Trifluoromethoxy)benzylidene]-N-[(4-methylthio)phenyl]hydrazinecarbo thioamide (IId). White solid (chloroform). Yield: 0.25 g (74%); m.p.: 198 – 199°C. IR (νmax, cm-1): 3324, 3121 (N-H); 1577 (C=N); 1160 (C-N); 1077 (N-N); 822 (C=S). 1H NMR (DMSO-d6; δ, ppm): 11.91 (s, 1H, CSNH); 10.15 (s, 1H, PhNH); 8.17 (s, 1H, HC=N); 8.06 (d, 2H, ArH, J = 8.57 Hz, meta protons to -SCH3 group); 7.51 (d, 2H, ArH, J = 8.55 Hz, meta protons to -OCF3 group); 7.42 (d, 2H, ArH, J = 8.52 Hz, orto protons to -OCF3 group); 7.28 (d, 2H, ArH, J = 8.56 Hz, orto protons to -SCH3 group); 2.49 (s, 3H, -SCH3). 13C NMR (DMSO-d6; δ, ppm): 176.40 (C=S), 147.36 (ArC, ipso carbon to -OCF3 group), 143.71 (C=N), 141.11, 139.78, 136.33, 129.81 (ArC), 127.04 (OCF3), 125.92, 122.49, 116.12 (ArC), 15.84 (SCH3).
2-(4-Fluorobenzylidene)-N-[(4-methylthio)phenyl]hydrazinecarbothioamide (IIe). White solid (chloroform). Yield: 0.24 g (72%); m.p.: 175 – 176°C. IR (νmax, cm-1): 3323, 3133 (N-H); 1581 (C=N); 1228 (C-F); 1150 (C-N); 1069 (N-N); 826 (C=S). 1H NMR (DMSO-d6; δ, ppm): 11.83 (s, 1H, CSNH); 10.11 (s, 1H, PhNH); 8.15 (s, 1H, HC=N); 7.99 (dd, 2H, ArH, J = 8.70 Hz, meta protons to -F atom); 7.51 (d, 2H, ArH, J = 8.57 Hz, meta protons to -SCH3 group); 7.28 (t, 2H, ArH, J = 8.77 Hz, orto protons to –F atom); 7.27 (d, 2H, ArH, J = 8.55 Hz, orto protons to -SCH3 group); 2.49 (s, 3H, -SCH3). 13C NMR (DMSO-d6; δ, ppm): 176.89 (C=S), 164.01 (ArC, ipso carbon to -F atom), 143.18 (C=N), 140.04, 133.95, 131.56, 130.19, 127.76, 119.31, 115.83 (ArC), 16.24 (SCH3).
2-(4-Nitrobenzylidene)- N -[(4-methylthio)phenyl]hydrazinecarbothioamide (IIf).
Orange solid (acetone). Yield: 0.30 g (82%); m.p.: 225 – 226°C. IR (νmax, cm-1): 3296, 3120 (N-H); 1587 (C=N); 1435 (NO2, asymmetric); 1336 (NO2, symmetric); 1185 (C-N); 1083 (N-N); 836 (C=S). 1H NMR (DMSO-d6; δ, ppm): 12.10 (s, 1H, CSNH); 10.30 (s, 1H, PhNH); 8.27 (d, 2H, ArH, J = 8.96 Hz, orto protons to -NO2 group); 8.24 (s, 1H, HC=N); 8.21 (d, 2H, ArH, J = 8.95 Hz, meta protons to -NO2 group); 7.50 (d, 2H, ArH, J = 8.58 Hz, meta protons to -SCH3 group); 7.29 (d, 2H, ArH, J = 8.59 Hz, orto protons to -SCH3 group); 2.50 (s, 3H, -SCH3). 13C NMR (DMSO-d6; δ, ppm): 176.57 (C=S), 149.15 (ArC, ipso carbon to -NO2 group), 142.86 (C=N), 140.54, 139.27, 133.66, 130.83, 129.11, 128.77, 126.35 (ArC), 15.88 (SCH3).
2-{4-[Bis(2-chloroethyl)amino]benzylidene}-N-[(4-methylthio)phenyl]hydrazine carbothioamide (IIg). Light yellow solid (chloroform). Yield: 0.40 g (88%); m.p.: 192 – 193°C. IR (νmax, cm-1): 3299, 3162 (N-H); 2228 1545 (C=N); 1177 (C-N); 1061 (N-N); 818 (C=S); 712 (C-Cl). 1H NMR (DMSO-d6; δ, ppm): 11.62 (s, 1H, CSNH); 9.92 (s, 1H, PhNH); 8.04 (s, 1H, HC=N); 7.72 (d, 2H, ArH, J = 8.82 Hz, meta protons to -SCH3 group); 7.55 (d, 2H, ArH, J = 8.58 Hz, meta protons to -N(CH2CH2Cl)2 group); 7.26 (d, 2H, ArH, J = 8.60 Hz, orto protons to -N(CH2CH2Cl)2 group); 6.80 (d, 2H, ArH, J = 8.90 Hz, orto protons to -SCH3 group); 3.75 – 3.82 (m, 8H, -N(CH2CH2Cl)2 protons); 2.50 (s, 3H, -SCH3). 13C NMR (DMSO-d6; δ, ppm): 175.96 (C=S), 148.41 (ArC, ipso carbon to -N(CH2CH2Cl)2 group), 143.89 (C=N), 136.70, 134.56, 129.72, 126.44, 126.12, 122.63, 112.06 (ArC), 52.32 (N(CH2CH2Cl)2), 41.87 (N(CH2CH2Cl)2), 15.57 (SCH3).
3. Experimental Biological Part
3.1. Antiviral Activity Assay
The antiviral assays were based on the inhibition of virus-induced cytopathicity in confluent cell cultures, and the cytostatic assays on inhibition of tumor cell proliferation in exponentially growing tumor cell cultures according to previously described methods [17]. The synthesized compounds were evaluated against the following viruses: herpes simplex virus type 1 (HSV-1) strain KOS, thymidine kinase-deficient (TK–) HSV-1 KOS strain resistant to ACV (ACVr), herpes simplex virus type 2 (HSV-2) strain G, cytomegalovirus strains AD-169 and David, varicella zoster virus (VZV) strains OKA and YS, vaccinia virus Lederle strain, respiratory syncytial virus (RSV) strain Long, vesicular stomatitis virus (VSV), Coxsackie B4 virus, parainfluenza 3 virus, influenza virus A (subtypes H1N1, H3N2), influenza virus B, reovirus-1, Sindbis and Punta Toro virus. The antiviral assays were based on inhibition of virus-induced cytopathicity or plaque formation (for VZV) in human embryonic lung (HEL) fibroblasts, African green monkey cells (Vero), human epithelial cervix carcinoma cells (HeLa) or Madin-Darby canine kidney cells (MDCK). Confluent cell cultures in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) or 20 or 100 plaque forming units (PFU) (for VZV and CMV, respectively) in the presence of varying concentrations of the test compounds. Viral cytopathicity or plaque formation was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as EC50 (50% effective concentration) defined as the compound concentration required to reduce virus-induced cytopathogenicity or viral plaque formation by 50%). Inhibition of HIV-1(IIIB)- and HIV-2(ROD)-induced cytopathicity in CEM cell cultures was measured in microtiter 96-well plates containing ~3 × 105 CEM cells/mL infected with 100 CCID50 of HIV per milliliter and containing appropriate dilutions of the test compounds. After 4 – 5 days of incubation at 37°C in a CO2-controlled humidified atmosphere, CEM giant (syncytium) cell formation was examined microscopically and characterized by the EC50 value (50% effective concentration inhibiting HIV-induced giant cell formation by 50%).
3.2. Cytostatic/Toxic Activity Assay
All cytostatic/toxic activity assays were performed in 96-well microliter plates. To each well were added (5 – 7.5) × 104 tumor cells and a given amount of the test compound. The cells were allowed to proliferate for 48 h (murine leukemia L1210 cells) or 72 h (human CD4+ T-lymphocytic CEM and human cervix carcinoma HeLa cells) at 37°C in a humidified CO2-controlled atmosphere. At the end of the incubation period, the cells were counted in a Coulter counter. The IC50 (50% inhibitory concentration) was defined as the concentration of the compound that inhibited cell proliferation by 50%. Cisplatin was purchased from SIGMA and used as a reference drug. Compound concentrations of 100, 20, 4, 0.8, 0.16 and 0.032 μg/mL have been tested (5-fold dilutions) and the data represent the mean ± SD (standard deviation) of at least two to three independent experiments. All values are significantly different from cisplatin (less active), except for Ig (no statistical difference, p > 0.05).
4. Experimental Molecular Modeling Part
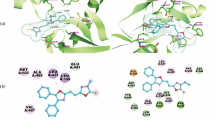
The biological activity depended on only on the chemical structure of substituents, but also on the three dimensional (3D) configuration of compound Ig. For this reason, we again applied computational methods and ligand-based pharmacophore modeling to evaluate this state. The new derivatives of thiosemicarbazones (Ia – Ig and IIa – IIg) were drawn and converted from 2D to 3D, and the PM3MM basis set was assigned and then minimized using a semi-empirical method with the aid of Gaussian09 program package. A conformational search of the ligands was carried out using BEST algorithm for 3D pharmacophore generation. The Auto Pharmacophore Generation protocol in Discovery Studio 3.5 was used based on a bioactive conformation. In this study, we used compound Ig as the bioactive conformation with IC50 values between 0.30 and 2.50 μg/mL. The pharmacophore models were generated based on the main features of compound Ig. These features were Hb-Acceptor, Hb-Donor, Hydrophobic and Ring Aromatic in the pharmacophore models. Besides, the Genetic Function Approximation (GFA) model for the selectivity of pharmacophores was used. The best model was determined with the help of Clusters pharmacophores subprotocol. The model contains six features, representing two hydrophobic (cyan), two ring aromatic (orange), one hydrogen bond donor (pink) and one hydrogen acceptor (green).
5. Results and Discussion
All newly synthesized compounds were evaluated for their antiviral, cytostatic, and cytotoxic properties. None of the compounds effectively inhibited DNA virus replication (i.e. HSV-1, HSV-2, VV), except I and II that showed slight activity against vaccinia virus replication in HEL cell cultures (EC50 = 45 – 50 μg/mL) and compounds Ib, Ic and IIc bearing an electron-donating group (–OH and –OCH3) against feline herpes virus (EC50 = 17.8 – 54.3 μg/mL). All compounds were also not significantly inhibitory against a wide variety of RNA viruses, including vesicular stomatitis virus (VSV), respiratory syncytial virus and Coxsackie virus in HeLa cell cultures at subtoxic concentrations (Table 2). However, in Vero cell cultures compound Ig consistently showed an antiviral activity of 3 μg/mL (EC50) against Coxsackie virus B4 but there was no inhibitory activity observed for parainfluenza virus-3, reovirus-1, Sindbis virus and Punta Toro virus (Table 2). The fact that the activity of Ig against Coxsackie virus B4 was only observed in one cell line (monkey kidney Vero), but not in another cell line (human cervix carcinoma HeLa) led us to conclude that this compound should not be considered as a consistent new antiviral lead agent.
A marginal activity was observed for Ib, Ic and IIc against feline herpes virus (EC50 = 17.8 – 54.3 μg/mL) and for Ib, Ic and IIa against feline corona (FIPV) virus (EC50 = 7.0 – 31.5 μg/mL) in CRFK cell cultures (Table 3). Whereas no activity was found in MDCK cell cultures against influenza B virus, compounds Id, Ie, If and IIf which have an electron-withdrawing group (–CF3, –F and –NO2) proved inhibitory against influenza A virus (EC50: 12.7 – 26.7 μg/mL for strain H1N1 and 9.3 – 18.5 μg/mL for strain H3N2). These compounds were only 5- to 20-fold less effective than the clinically used drug oseltamivir and were neither cytotoxic (MCC) nor cytostatic (CC50) at 100 μg/mL against MDCK cells (Table 3). Thus, a certain degree of selectivity was present for these compounds against influenza A virus in cell culture and should be further considered for optimization as potential anti-influenza virus lead compounds.
The synthesized compounds were also evaluated for their cytostatic activity against murine leukemia L1210, human CD4+ T-lymphocyte CEM and cervix carcinoma HeLa cells (Table 4). Most compounds in the IIa – IIg series did not show pronounced cytostatic activity (IC50 ≥ 100 μg/mL). However, IIb was moderately cytostatic (IC50 = 13 – 15 μg/mL), whereas IIg seemed to be poorly inhibitory to L1210 and CEM cell proliferation, but had a pronounced antiproliferative activity against HeLa cells (IC50 = 1.9 μg/mL). This unusual selectivity between tumor cell lines should be further investigated. In fact, the I series of compounds were often somewhat more cytostatic than their corresponding II series of compounds. It may not be a coincidence that Ig (IC50 = 0.3 μg/mL against HeLa cells) was also most cytostatic among series I and also showed the most pronounced antiproliferative potency against HeLa cells.
The structure – activity relationship (SAR) studies were performed to determine how the substituent on the benzene ring affected the cytostatic activity. Compounds Ia and IIa without any substituent group on the benzene ring did not show cytostatic activity. Then, we examined the effect of introducing various substituent groups in the benzene ring of compounds Ia and IIa. The -OH, -OCH3, -CF3, -F, -NO2 and -N(CH2CH2Cl)2 groups at the para position of the benzene ring of compounds Ia and IIa were used to form compounds (Ib – Ig and IIb – IIg). Especially Ig with a bis(2-chloroethyl) amino moiety, the IC50 values (0.30 – 2.50 μg/mL) were the lowest (most effective) among all compounds and compared to cisplatin as a reference compound. Atwo-tailed, unpaired t-test (GraphPad Prism) revealed no significant difference between the cytostatic inhibition values of Ig and cisplatin obtained against the three tumor cell lines (p = 0.647). The results showed that compounds Ia – Ig containing electron-withdrawing group, such as -CN located at the para position of the benzene ring, increased the cytostatic activity, according to the electron-donating group (-SCH3) at the same position of the structure (like compounds IIa – IIg). Furthermore, the introduction of electron-donating groups (-OH, -OCH3) at the para position of the benzene ring weakened the cytostatic effects. While the same situation for electron-withdrawing groups (-F, -NO2,-CF3) was also observed, compound IIb exhibited moderate activity against all tumor cell types. Besides, compound 1g displayed the better cytostatic activity than its analog compound IIg, which has an electron-donating group (-SCH3) at the para position. These results indicated that the introduction of the bulky alkylating group (bis(2-chloroethyl)amino) and a cyano group reinforced the cytostatic activity.
The pharmacophore model (Fig. 2A) was hypothetically superimposed with compounds Ig (fit value 4.6216) and IIg (fit value 3.0032). Figures 2B and 2C summarize the results of model fitting obtained for compounds Ig and IIg. The other compounds were not superimposed to fit with the best model. In addition to comparing the results of modeling with experimental data, they show compatible and significant trends. It is established that Hb-acceptor (-CN; -SCH3), hydrophobic groups (-N(CH2CH2Cl)2), and 3D-conformations are responsible for the cytostatic activity of compounds such as Ig and IIg, according to ligand-based pharmacophore modeling. Especially, 3D-conformation of the investigated compounds showed the major effect in this study. Unfortunately, compounds Ia – If and IIa – IIf did not show any activity in comparison to compounds Ig and IIg. The proposed methods were able to provide valuable information about key features and interactions that are important for the biological activity of compound Ig. Thus, ligand-based pharmacophore modeling was increasingly successful in delineating why compound Ig displayed better cytostatic activity than compound IIg.

(A) Pharmacophore model of the docking pose of compound Ig; (B) the mapping of compound Ig (fit value 4.6216) to hypo10; (C) the mapping of compound IIg (fit value 3.0032) to hypo10.
References
G. Domagk, R. Behnisch, F. Mietzsch, et al., Naturwissenschaften, 33, 315 (1946).
D. Hamre, J. Bernstein. and R. Donovick, Proc. Soc. Exp. Biol. Med., 73, 275 – 278 (1950).
D. J. Bauer, L. St. Vincent, C. H. Kempe, et al., Lancet, 2, 494 – 496 (1963).
M. A. Soares, J. A., Lessa, I. C. Mendes, et al., Bioorg. Med. Chem., 20, 3396 – 3409. (2012).
R. A. Finch, M.-C. Liu, S. P. Grill, et al. Biochem. Pharmacol., 59, 983 – 991 (2000).
V. R. Solomon, C. Hu, and H. Lee, Bioorg. Med. Chem., 18, 1563 – 1572 (2010).
D. M. Noll, T. M. Mason, and P. S. Miller, Chem. Rev., 106, 277 – 301 (2006).
F. F. Fleming, L. Yao, P. C. Ravikumar, et al. J. Med. Chem., 53, 7902 – 7917 (2010).
A. Karaküçük-Iyidoğan, D. Taşdemir, E. E. Oruç-Emre, et al., Eur. J. Med. Chem., 46, 5616 – 5624 (2011).
M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al. Gaussian 09, Revision A.1 Gaussian, Inc., Wallingford CT (2009).
Accelrys Software Inc., Discovery Studio 3.5, San Diego (2013).
A. Karaküçük-Iyidoğan, Z. Mercan, E. E. Oruç-Emre, et al. Phosphorus Sulfur and Silicon, 189, 661 – 673 (2014).
H. Huang, Q. Chen, X. Ku, et al., J. Med. Chem., 53, 3048 – 3064 (2010).
M. Leigh, D. J. Raines, C.E. Castillo, et al. Chem. Med. Chem., 6, 1107 – 1118 (2011).
A.-C. Tnechiu (Deleanu), I. D. Kostas, D. Kovala-Demertzi, et al. Carbohydr. Res., 344, 1352 – 1364 (2009).
V. A. Chornous, A. N. Grozav, L. D. Todoriko, et al. Pharm. Chem. J., 47(10), 524 – 526 (2014).
E. Tsoukala, N. Tzioumaki, S. Manta, et al. Bioorg. Chem., 38, 285 – 293 (2010).
Acknowledgements
The present work was supported by the Scientific Research Projects Governing Unit Council of Gaziantep University (Grant No. FEF.08.10), Gaziantep, Turkey and by the KU Leuven (GOA 10/14). The authors are grateful to Leentje Persoons, Frieda De Meyer, Leen Ingels, Anita Camps, Steven Carmans, Lies Van den Heurck and Lizette van Berckelaer for excellent technical assistance and Dr. Annelies Stevaert for statistical analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Karaküçük-İyidoğan, A., Aydınöz, B., Taşkın-Tok, T. et al. Synthesis, Biological Evaluation and Ligand Based Pharmacophore Modeling of New Aromatic Thiosemicarbazones as Potential Anticancer Agents. Pharm Chem J 53, 139–149 (2019). https://doi.org/10.1007/s11094-019-01968-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-019-01968-3