
Abstract
Methane conversion and in particular the formation of the C-O bond is one of fundamental entries to organic chemistry and it appears to be essential for understanding parts of atmospheric chemistry of Titan, but, in broader terms it might be also relevant for Earth-like exoplanets. Theoretical study of the reactions of methane with atomic oxygen ion in its excited electronic states requires treating simultaneously at least 19 electronic states. Development of a computational strategy that would allow chemically reasonable and computationally feasible treatment of the CH4 (X)/O+ (2D, 2P) system is by far not trivial and it requires careful examination of all the complex features of the corresponding 19 potential energy surfaces. Before entering the discussion of the rich (photo) chemistry, inspection of the long range behavior of the system with focus on electric dipole transition moments is required. Our calculations show nonzero probability for the reactants to decay before entering the multiple avoided crossings region of the [CH4 + O → products]+ reaction. For the CH4/O+ (2P) system non-zero transition moment probabilities occur over the entire range of considered C-O distances (up to 15 Å), while for the CH4/O+ (2D) system these probabilities are lower by one order of magnitude and were found only at C-O distances smaller than 6 Å.
Similar content being viewed by others
Introduction
Titan provides an extraordinary environment to better understand some of the chemical processes that contributed to the apparition of life on Earth. Its atmosphere is a natural laboratory that, in many aspects, seems to have a partial similitude with our current picture of the pre-biotic atmosphere of Earth. The research paper, “Clues on the importance of comets in the origin and evolution of the atmospheres of Titan,” by Trigo-Rodriguez and Martín-Torres (2012) offers insight into the atmospheric affinities of Earth and Titan. The presence of oxygen atoms on Titan (Hörst et al. 2008; Moreno et al. 2012) and its ions (Cravens et al. 2008) potentially provides the basis for pre-biological chemistry that could be helpful for explaining early Earth’s organic chemical evolution. Therefore, it appears very important to fully understand the reactions of O+ with the two most abundant neutrals on Titan, namely N2 and CH4 (Dobrijevic et al. 2014). This goal encouraged already numerous experimental and theoretical works (see, e.g., (Cunha de Miranda et al. 2015 and refs. therein).
The reaction mechanism and in particular the dynamics and kinetics of the underlying processes are not known till now in full detail, despite many studies performed on this system. The problem got further impetus in the context of recent debate on the role of oxygen in Titan atmosphere (Hörst et al. 2008). It was indicated that excited O+ ions could be the starting point of chemistry and that O+ reactions with N2 and CH4 represent the entry to rich palette of organic (“prebiotic”) molecules. The reactions of O+(4S, 2D, 2P) with N2 were in detail investigated (Li et al. 1997). Complementary experiments on the state-selected O+(4S, 2D, 2P) atomic ions with CH4 (and CD4) were performed using the CERISES setup at synchrotron SOLEIL in Paris (Cunha de Miranda et al. 2015). Absolute O+/CH4 (O+/CD4) cross sections were measured at selected collision energies. For the ground state and the excited states a reduction of the total cross section and a complete inversion of the branching ratio between the main products (CH4 + and CH3 +) in favor of the latter is observed.
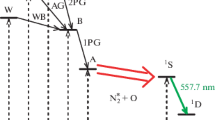
We focus on the O+ + CH4 reaction (Hrušák and Paidarová 2013) with particular emphasis on the excited states reactivity. Contrary to the reaction of methane with O+(4S) ground state, more products become energetically accessible when including the O+(2D, 2P) reactants, as it is seen from the table in Fig. 1. Understanding of the richness of the reaction products and the underlying reaction mechanism benefits from the interplay of experimental and theoretical techniques. Already the very first ion-molecule reaction studies using the guided ion beam (GIB) technique have to be assisted by theoretical ab initio calculations of stationary points on potential energy surfaces (PES’s) (Li et al. 1997; Levandier et al. 2004) and by dynamic studies of reactions of the ground state ion O+(4S) by (Sun and Schatz 2005). Further understanding was gained by a combined experimental/theoretical study of chemical reactivity aiming to improve astrochemistry models, e.g. (Krasnopolsky 2012, 2014). Finally, experiments, which indicate that different reactions channels may be acting for the individual O+ electronic states were performed recently (Cunha de Miranda et al. 2015). This all is the motivation for our investigations by means higher level theoretical approaches.

Energy diagram for the thermodynamically accessible reaction products of the O+/CH4 collision constructed from accessible data. All energies are in eV relative to the CH4/O+(4S) entry that is set 0.0 eV. The individual electronic states are indicated where ever it is relevant (X - denotes the ground state and A, B, C are the corresponding excited states)
The complexity of the system does not allow traditional dynamic studies as they were performed for the O+(4S) + CH4 (i.e. the ground state) reaction. Figure 1 indicates the states of the (O/CH4) +/0 system to be considered. In order to facilitate the reactions of O+(2D) + CH4 and of O+(2P) + CH4, the symmetry cannot be reduced below Cs. That leads to the necessity to treat in parallel at least 19 states of A’ irreducible representation to encompass the minimal energy window. Such calculations are extremely demanding and bounce the technical limits of recent computational codes. A feasible approach, called ”chemically reasonable intuitive reaction coordinate” (Hrušák and Paidarová 2013), dramatically reduces the dimensionality of the calculations required for the (O/CH4)+ system. It replaces the traditional multidimensional equidistant grid calculations of the PES’s by a series of interconnected cuts, along which the positions of avoided crossings are located and all the properties are followed.
Experiments suggest (Cunha de Miranda et al. 2015) that charger transfer and dissociative charge transfer are dominant reactions (i.e. CH4 + and CH3 + being the main products). These, processes typically occur in distant intermolecular regions larger than 5 Å (cf. Closs and Miller 1988; Frey et al. 2013), i.e. well before chemical interactions can become active. Thus, it appears useful to closer inspect the long range distance behavior of the above mentioned reaction with focus on electric dipole transition moments.
An additional feature, which is immediately apparent from last column in Fig. 1, is the very broad pallet of organic molecules and reactive molecular fragments. All these systems are energetically accessible within the energy window. Work is in progress that enables to discuss important product channels, in particular those leading to the creation of the C-O bond. In addition, once formed, these species open, through a series of subsequent collisions with nitrogen, straight pathways to prebiotic molecules including amino-acids. However, it is still a long way to go, before these studies will impact sizably the present atmospheric models.
Calculations
The calculations of energies and transition dipole moments reported here, were performed by applying the multi-configurational self-consistent field (MCSCF) approach (Werner and Knowles 1985; Knowles and Werner 1985) in conjunction with the augmented correlation consistent triple zeta basis set (aug-cc-pVTZ). In the MCSCF calculation all orbitals were fully relaxed and optimized. In order to access all the states required (in particular those containing the CH4 (1A1) + O+ (2D, 2P) asymptotes) Cs symmetry group was used throughout all the calculations. On the doublet surfaces 19 lowest electronic states of A’ irreducible representation with equal weights were considered in the state-averaged (SA) MCSCF (SA-MCSCF) calculations. In the same manner 16 A” states were taken into account. It was shown previously (Hrušák and Paidarová 2013) that CH3 + can be formed by a chemical reaction occurring at C-O distances around 2 Å. Thus, here we concentrate on the long range region presented in Fig. 2, where the CH4 + could be formed by a charge transfer process and, subsequently, the CH3 + would appear as a result of vibrational energy redistribution of the hot CH4 + (so called dissociative charge transfer). The corresponding part of the PES’s is drawn as a function of OX and AX coordinates, where OX describes O-CH4 distance and AX designates the impact parameter. For OX >5 a.u. and AX >4 a.u. the PES’s are flat, degenerated and quasi-parallel.

Cut through the potential energy surfaces corresponding to 19 A’ states of CH4/O+ as functions of OX and AX coordinates
The charge transfer between the electronic states i, j is controlled by the square value of the electric dipole transition moments
where ψ i , ψ j are electronic wave functions of the states i and j. Asymptotically degenerate states corresponding to CH4 (1A1) + O+ (2D) and CH4 (1A1) + O+(2P), are the 14th and 15th, and 16th and 17th state, respectively. In order to get a qualitative picture about the electric dipole transition moments behavior we have summed up the
and
and the results are shown in Fig. 3

Square value of the electric dipole transition moments (atomic units) from the states corresponding to asymptotic limit O+(2D) + CH4 and O+(2P) + CH4, a and b respectively, to all available lower states of (CH4/O)+ as functions of the nuclear coordinates OX and AX
As shown in Fig. 2, the energy profile of all the PESs is rather flat in the OX/AX long range area. The coupling of several states around 10 Å remains to be understood. However, even after this discontinuity the energy profiles remain quasi-parallel. Significant interactions, i.e. the chemistry, occur only at very small OX/AX distances (bellow 3.5 Å).
The investigation of the electric dipole transition moments between the states corresponding to the reactants O+ (2D, 2P) + CH4 (1A1) and all the energetically lower states aims to answer the question important for the subsequent dynamic studies: Do the excited O+ ions under single collision conditions reach the “chemically relevant” avoided crossings region (short AO/OX distances), or does the charge transfer prevailingly occur in the long-range (above 5 Å) part of the surfaces? The corresponding tm surfaces are shown in Fig. 3. The probability of radiative charge transfer at the long range is nonzero for CH4 (1A1) + O+ (2P) states, while for the lower excited state of oxygen (2D) the probability of radiative decay is very small and almost zero in the long range part of the nuclear configurations. This has immediate consequences for the lifetimes and for the reactivity. The O+ (2P) decays faster as its lower lying electronic counterpart (2D) and thus, it is to lesser extent available for subsequent chemical reactions. Therefore, regardless of the fact that the (2P) oxygen ion carries more potential energy, its significance for overall product distribution is much lower compared to the O+ (2D) ion, of which larger fraction survives to the region where photochemistry can occur. One could also speculate, that for the 2P state of oxygen the subsequent chemistry is dominated by the CH4 + behavior, due to charge transfer occurring at longer distances. Thus, and in line with experimental observations, differences in reactivity and in product distributions can be expected between these two states of oxygen ion reacting with methane. It must be emphasized, however, that all these considerations are valid for single collision conditions only and it must be proven by dynamic studies.
Concluding Remarks
The energetics of the possible reaction products is shown in the Fig. 1. The energy window (green rectangle in the 2nd column) encompasses the three states of atomic oxygen up to the energetically high lying channel (CH4 + O+(2P)). In (Hrušák and Paidarová 2013) we have focused on charge transfer and/or OH elimination reaction, which are the principal reaction products as was found in the recent experiments (Cunha de Miranda et al. 2015). No principle differences were found for the CH3 + formation irrespectively of the electronic state of the reactants. On the contrary, when inspecting the long range behavior of the transition moments clear differences in behavior are observed for the two excited states of O+. In that sense this study represents a step towards the extension of the CRIRC model (Hrušák and Paidarová 2013) in preparation of multi-state multi-level approach to dynamics. In addition, up to now we included at the product site only the bound electronic states of OH, OH+, CH3, CH3 +, and CH4 + and we did not consider excited states for the secondary reaction products CH3O+, CH2O+, HCO+, and COH+ etc. (blue rectangle in the 5th column in Fig.1). Even though, these species represent only a small fraction of the thermal reaction products, they are essential for the reactions leading to the creation of more complex molecules, i.e. building elements of the prebiotic processes. Complete theoretical description of the [CH4/O]+ chemistry is required, before these studies will contribute to the improvements of existing photochemical models of Titans atmosphere (Lara et al. 2014).
References
Closs RL, Miller JR (1988) Intramolecular long distance electron transfer in organic molecules. Science 240(4851):440
Cravens TE, Robertson IP, Ledvina SA, Mitchel D, Krimings SM, Waite JH Jr. (2008) Energetic ion precipitation at titan. Geophys Res Lett 35:L03103
Cunha de Miranda B, Romanzin C, Chefdeville S, Vuitton V, Žabka J, Polášek M, Alcaraz C (2015) Reactions of state-selected atomic oxygen ions O+(4S, 2D, 2P-2) with methane. J Phys Chem A 119:6082–6098
Dobrijevic M, Hébrard E, Loison JC, Hickson KM (2014) Coupling of oxygen, nitrogen, and hydrocarbon species in the photochemistry of Titan’s atmosphere. Icarus 228:324
Frey J, Kodis G, Straight SD, Moore TA, Moore AL, Gust D (2013) Photonic modulation of electron transfer with switchable phase inversion. J Phys Chem A 117:607
Hörst SM, Vuitton V, Yelle RV (2008) Origin of oxygen species in Titan’s atmosphere. J Geophys Res 113:E10006
Hrušák J, Paidarová I (2013) The way toward theoretical description of state-selected reactions of O+ with methane. Int J Mass Spectrom 354:372–377
Knowles PJ, Werner H-J (1985) An efficient second-order MCSCF method for long configuration expansions. Chem Phys Lett 115:259
Krasnopolsky VA (2012) Titan’s photochemical model: further update, oxygen species, and comparison with triton and Pluto. Planet Space Sci 73:318–326
Krasnopolsky VA (2014) Chemical composition of Titan’s atmosphere and ionosphere: observations and the photochemical model. Icarus 236:83–91
Lara LM, Lellouch L, González M, Moreno R, Rengel M (2014) A time–dependent photochemical model for Titan’s atmosphere and the origin of H2O. Astron Astrophys 566:A143
Levandier JD, Chiu Y-H, Dressler RA, Sun L, Schatz GC (2004) Hyperthermal reactions of O+(4S3/2) with CD4 and CH4: theory and experiment. J Phys Chem A 108:9794–9804
Li X, Huang YL, Flesch GD, Ng CY (1997) A state-selected study of the ion-molecule reactions O+(4S, 2D, 2P) + N2. J Chem Phys 106:1373
Moreno R, Lellouch E, Lara LM, Feuchtgruber H, Rengel M, Hartogh P, Courtin R (2012) The abundance, vertical distribution and origin of H2O in Titan’s atmosphere: Herschel observations and photochemical modelling. Icarus 221:753
Sun L, Schatz GC (2005) Direct dynamics classical trajectory simulations of the O+ + CH4 reaction at Hyperthermal energies J. Phys Chem B 109:8431–8438
Trigo-Rodriguez JM, Martín-Torres FJ (2012) Clues on the importance of comets in the origin and evolution of the atmospheres of titan and earth. Planet Space Sci 60:3
Werner H-J, Knowles PJ (1985) A second order multiconfiguration SCF procedure with optimum convergence. J Chem Phys 82:5053
Acknowledgments
This work results within the collaboration of the COST Action TD 1308.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hrušák, J., Paidarová, I. Step Towards Modeling the Atmosphere of Titan: State-Selected Reactions of O+ with Methane. Orig Life Evol Biosph 46, 419–424 (2016). https://doi.org/10.1007/s11084-016-9503-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-016-9503-4