
Abstract
The first Principle calculations are made to study the structural electronic and optical properties of indium-doped aluminum antimonide. The most appropriate method of density functional theory (DFT) naming Full Potential Linearized Augmented Plane Wave (FP-LAPW) is used. The structural properties like Lattice constant (a), pressure derivative, and bulk modulus (B) of Al1−xInxSb (x = 0, 0.25, 0.5, 0.75) are examined with generalized gradient approximation (GGA). Generalized gradient approximation along with TB-mBJ is used to determine electronic parameters like band structure along and density of states. According to the computed results the binary compound AlSb is optically inactive and exhibits an indirect (Γ-L) band gap. By increasing the concentration of indium with different percentages, the indirect band gap shifted to the direct (Γ-Γ) band gap which shows the material is optically active. The optical properties of the material including dielectric (Real and imaginary parts) constant, reflectivity, refractive index, energy loss, absorption coefficient, and optical conductivity have changed significantly. Electronic and optical properties are modified by (TB-mBJ) approach. The results obtained are examined with experimental data and utilized as a starting point to propose that the material is the superlative choice for the manufacturing of p-n junctions, photo-detectors, laser, photo-diodes, transistors and solar spectrum absorptions in the visible, infrared and ultraviolet energy ranges.
















Similar content being viewed by others
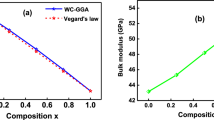
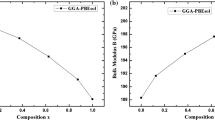
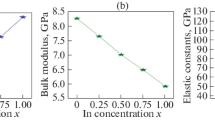
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Abbassi, A., Zaari, H., Azahaf, C., Ez-Zahraouy, H., Benyoussef, A.: Spontaneous polarization and magnetic investigation of BiXO3 (X = Co, Mn, Fe, V, Zn): first-principle study. J. Supercond. Nov. Magn. 29(2), 487–491 (2016)
Ahmed, R., Hashemifar, S.J., Akbarzadeh, H., Ahmed, M., Fazal-e-Aleem: Ab initio study of structural and electronic properties of III-arsenide binary compounds. Comput. Mater. Sci. 39(3), 580–586 (2007)
Ahmed, R., Fazal-e-Aleem, Hashemifar, S.J., Akbarzadeh, H.: First-principles study of the structural and electronic properties of III-phosphides. Phys. B Condens. Matter 403(10–11), 1876–1881 (2008)
Ali, Z., Khan, I., Rahman, M., Ahmad, R., Ahmad, I.: Electronic structure of the LiAA′O6 (A = Nb, Ta, and A′ = W, Mo) ceramics by modified Becke-Johnson potential. Opt. Mater. 58, 466–475 (2016)
Ali, M.A., Khan, N., Ahmad, F., Ali, A., Ayaz, M.: First-principles calculations of opto-electronic properties of IIIAs (III = Al, Ga, In) under influence of spin–orbit interaction effects. Bull. Mater. Sci. 42(1), 1–10 (2019)
Ali, M.A., Aleem, H., Sarwar, B., Murtaza, G.: First-principles calculations for optoelectronic properties of AlSb and GaSb under influence of spin–orbit interaction effect. Indian J. Phys. 94(4), 477–484 (2020)
Al-zyadi, J.M.K., Asker, H.I., Yao, K.: electronic structure and magnetic properties of the (111), (110) and (001) surfaces for the full heusler alloy Zr2VGa. J. Supercond. Nov. Magn. (2020). https://doi.org/10.1007/s10948-020-05654-4
Ameri, M., Bennar, F., Amel, S., Ameri, I., Al-Douri, Y., Varshney, D.: Structural, elastic, thermodynamic and electronic properties of LuX (X = N, Bi and Sb) compounds: first principles calculations. Ph. Transit. 89(12), 1236–1252 (2016)
Amin, B., Ahmad, I., Maqbool, M., Goumri-Said, S., Ahmad, R.: Ab initio study of the bandgap engineering of Al1−xGaxN for optoelectronic applications. J. Appl. Phys. 109, 2 (2011)
Annane, F., Meradji, H., Ghemid, S., Bendjeddou, H., EL Haj Hassan, F., Vipul Srivastav Khenata, R.: Ab-initio study of ordered III–V antimony-based semiconductor alloys GaP1−xSbx and AlP1−x Sbx. Pramana - J. Phys. 94(1), 1–16 (2020)
Baird, O., Wang, B., Wang, J., Chen, S,G., Lee, Y,S., Donadio, S., Kovnir, D., Kirill.: III-V clathrate semiconductors with outstanding hole mobility: Cs8In27Sb19 and A8Ga27Sb19 (A = Cs, Rb). J. Am. Chem. Soc. 142(4), 2031–2041 (2020)
Benchehima, M., Abid, H., Sadoun, A., Chabane Chaouche, A.: Optoelectronic properties of aluminum bismuth antimony ternary alloys for optical telecommunication applications: first principles calculation. Comput. Mater. Sci. 155, 224–234 (2018)
Bencherif, B., Abdiche, A., Moussa, R., Khenata, R., Wang, X.: Pressure effect on structural, electronic optical and thermodynamic properties of cubic AlxIn1–xP: a first-principles study. Mol. Phys. (2019). https://doi.org/10.1080/00268976.2019.1608380
Benkaboua, M.H., Harmela, M., Haddou, A., Yakoubi, A., Baki, N., Ahmed, N., Al-Douri, Y., Syrotyuke, S.V., Khachai, H., Khenata, H., Voong, C.H., Johanc, M.R.: Structural, electronic, optical and thermodynamic investigations of NaXF3 (X = Ca and Sr): first-principles calculations. Chinese J. Phys. 56(1), 131–144 (2018)
Benkaddour, Y., Abdelaoui, A., Yakoubi, A., Khachai, H., Al-Douri, Y., Bin Omran, S., Shankar, A., Khenata, R., Voon, C.H., Prakash, D., Verma, K.D.: First-principle calculations of structural, elastic, and electronic properties of intermetallic rare earth R2Ni2Pb (R = Ho, Lu, and Sm) compounds. J. Supercond. Nov. Magn. 31(2), 395–403 (2018)
Bennacer, H., Boukortt, A., Meskine, S., Hadjab, M., Ziane, M.I., Zaoui, A.: First principles investigation of optoelectronic properties of ZnXP2 (X = Si, Ge) lattice matched with silicon for tandem solar cells applications using the mBJ exchange potential. Optik (stuttg) 159, 229–244 (2018)
Bentayeb, A., Driss Khodja, F., Chibani, S., Marbouh, N., Bekki, B., Khalfallah, B., Elkeurti, M.: Structural, electronic, and optical properties of AlNxSb1−x alloys through TB–mBJ–PBEsol: DFT study. J. Comput. Electron. 18(3), 791–801 (2019)
Berrah, S., Boukortt, A., Abid, H.: Optical properties of the cubic alloy (In, Ga)N. Phys. E Low-Dimens. Syst. Nanostructures 41(4), 701–704 (2009)
Boudiaf, K., Bouhemadou, A., Al-Douri, Y., Khenata, R., Bin-Omran, S., Guechi, N.: Electronic and thermoelectric properties of the layered BaFAgCh (Ch = S, Se and Te): first-principles study. J. Alloys Compd. 759, 32–43 (2018)
Degheidy, A.R., Abdel, S., Elwakil, A., Elkenany, E.B.: Energy band structure calculations of GaxIn1−xP alloys under the influence of temperature and pressure. J. Alloys Compd. 574, 580–590 (2013)
Dimroth, F.: High-efficiency solar cells from III-V compound semiconductors. Phys. Status Solidi C: Conf. 3(3), 373–379 (2006)
El Haj Hassan, F., Breidi, A., Ghemid, S., Amrani, B., Meradji, H., Pagès, O.: First-principles study of the ternary semiconductor alloys (Ga,Al)(As,Sb). J. Alloys Compd. 499(1), 80–89 (2010)
Elkenany, E.B.: Theoretical investigations of electronic, optical and mechanical properties for GaSb and AlSb semiconductors under the influence of temperature. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 150, 15–20 (2015)
Erum, N., Iqbal, M.A.: Study of pressure variation effect on structural, opto-electronic, elastic, mechanical, and thermodynamic properties of SrLiF3. Phys. B Condens. Matter 525, 60–69 (2017)
Fahad, S., Murtaza, G., Ouahrani, T., Khenata, R., Yousaf, M., Bin Omran, S.: Structural, elastic, electronic, bonding and optical properties of BeAZ2 ( A = Si Ge Sn Z = P As ) chalcopyrites. J. Alloys Compd. 646, 211–222 (2015)
Finlayson, C.E., Ginger, D.S., Marx, E., Greenham, N.C., Ridley, B.K., Dobson, P.: Electrical and optical properties of semiconductor nanocrystals,. Philos. Trans. R. Soc. Lond. Ser. a: Math. Phys. Eng. Sci. 361(1803), 363–377 (2003)
Gazhulina, A.P., Marychev, M.O.: Structural, electronic and nonlinear optical properties of B3 and B20 compounds: a first-principles investigation within the LDA, GGA and modified Becke-Johnson exchange potential plus LDA. J. Alloys Compd. 623, 413–437 (2015)
Guemou, M., Abdiche, A., Riane, R., Khenata, R.: Ab initio study of the structural, electronic and optical properties of BAs and BN compounds and BNxAs1−x alloys. Phys. B Condens. Matter 436, 33–40 (2014)
Hasni, L., Ameri, M., Bensaid, D., Ameri, I., Mesbah, S., Yarub Al-Douri, Y., Coutinho, J.: First-principles calculations of structural, magnetic electronic and optical properties of rare-earth metals TbX (X = N, O, S, Se). J. Supercond. Nov. Magn. 30(12), 3471–3479 (2017)
Hattabi, I., Abdiche, A., Semari, F., Khenata, R., Soyalp, F.: Pressure effect on the structural, electronic and optical properties of the cubic triangular quaternary BNxAsyP1–x−y alloys: first principle study. Chin. J. Phys. 56(5), 2332–2349 (2018)
Hoat, D.M., Rivas Silva, J.F., Méndez Blas, A.: First principles study on structural, electronic and optical properties of Ga1−xBxP ternary alloys (x = 0, 0.25, 0.5, 0.75 and 1). Phys. Lett. Sect. A Gen. at. Solid State Phys. 382(29), 1942–1949 (2018)
Iqbal, R., Khan, I., Aliabad, H.A.R., Ali, Z., Ahmad, I.: Journal of magnetism and magnetic materials density functional studies of magneto-optic properties of CdCoS. J. Magn. Magn. Mater. 351, 60–64 (2014)
Joshi, T.K., Pravesh, Sharma, G., Verma, A.S.: Investigation of structural, electronic, optical and thermoelectric properties of Ethylammonium tin iodide (CH3CH2NH3SnI3): an appropriate hybrid material for photovoltaic application. Mater. Sci. Semicond. Process. (2020). https://doi.org/10.1016/j.mssp.2020.105111
Kafi, A., Khodja, F.D., Saadaoui, F., Chibani, S., Bentayeb, A., Khodja, M.D.: Materials science in semiconductor processing structural, elastic, electronic and thermoelectric properties of AlxGa1–xN(x. Mater. Sci. Semicond. Process. 113, 105049 (2020)
Khan, K., Gaur, A., Ahuja, U., Soni, A., Sahariya, J.: Density functional investigations to study effect of M = (Ge, Sn) doping on opto-electronic response of ZnSi(1–x)MxP2. Optik (stuttg) (2020). https://doi.org/10.1016/j.ijleo.2020.164570
Liu, W., Zheng, W.T., Jiang, Q.: First-principles study of the surface energy and work function of III-V semiconductor compounds. Phys. Rev. B – Condens. Matter. Mater. Phys. 75(23), 1–8 (2007)
Ma, C.G., Brik, M.G.: Hybrid density-functional calculations of structural, elastic and electronic properties for a series of cubic perovskites CsMF3 (M = Ca, Cd, Hg, and Pb). Comput. Mater. Sci. 58, 101–112 (2012)
Mao, Y., Liang, X.X., Zhao, G.J., Song, T.L.: Physica B : condensed matter the structural and optical properties of ternary mixed crystals InxGa1–xAs with zinc-blende structure by first-principle calculations. Phys. B Phys. Condens. Matter 569, 87–95 (2019)
Mbilo, M., Musembi, R.: First-principle calculations to investigate structural, electronic, elastic, mechanical, and optical properties of K2CuX (X=As, Sb) ternary compounds. Adv. Mater. Sci. Eng. (2022). https://doi.org/10.1155/2022/1440774
Moussa, R., Abdiche, A., Khenata, R., Soyalp, F.: First principles calculation of the structural, electronic, optical and elastic properties of the cubic AlxGa1−xSb ternary alloy. Opt. Mater. (amst) (2021). https://doi.org/10.1016/j.optmat.2021.110850
Murtaza, G., Ahmad, I.: First principle study of the structural and optoelectronic properties of cubic perovskites CsPbM3 (M = Cl, Br, I). Phys. B Condens. Matter 406(17), 3222–3229 (2011)
Nakano, K., Sakai, T.: Assessing the performance of the Tran-Blaha modified Becke-Johnson exchange potential for optical constants of semiconductors in the ultraviolet-visible light region. J. Appl. Phys. 123(1), 015104 (2018)
Perdew, J.P., Ruzsinszky, A., Csonka, G.I., Vydrov, O.A., Scuseria, G.E., Constantin, L.A., Zhou, X., Burke, K.: Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 13 (2008)
Rahman, G., Cho, S., Hong, S.C.: Half metallic ferromagnetism of Mn doped AlSb: a first principles study. Phys. Status Solidi Basic Res. 244(12), 4435–4438 (2007)
Rehman, G., Shafiq, M., Saifullah., Ahmad, R., Jalali-Asadabadi, S., Maqbool., Khan, M.I., Rahnamaye-Aliabad, H., Ahmad, I.: Electronic band structures of the highly desirable III–V semiconductors: TB-mBJ DFT studies. J. Electron. Mater. 45(7), 3314–3323 (2016)
Rodríguez-Hernández, P., Muñoz, A., Mujica, A.: High pressure phases of AlSb from ab-initio theory. Phys. Status Solidi Basic Res. 198(1), 455–459 (1996)
Rui, X., Hongbo, Y.,Yixuan, P., Bing, L., Ke, Y., Jiyang, L., Xiaolan, L.: Preparation of AlSb film by screen printing and sintering method. J. Mater. Sci. Mater. Electron. 30(14), 13290–13296 (2019)
Salehi, H., Allah, H., Farbod, M.: Materials science in semiconductor processing first principle study of the physical properties of semiconducting binary antimonide compounds under hydrostatic pressures. Mater. Sci. Semicond. Process. 26, 477–490 (2014)
Souadia, Z., Bouhemadou, A., Khenata, R., Al-Douri, Y.: Structural, elastic and lattice dynamical properties of the alkali metal tellurides: first-principles study. Phys. B Condens. Matter 521, 204–214 (2017)
Strössner, K., Ves, S., Koo Kim, C., Cardona, M.: Dependence of the direct and indirect gap of AlSb on hydrostatic pressure. Phys. Rev. B 33(6), 4044–4053 (1986)
Touam, S., Belghit, R., Mahdjhoubii, R., Megdoud, Y., Meradji, H., Shehryar Khan, M., Ahmed, R., Khenata, R., Ghemid, R., Rai, D.P., Al-Douri, Y.: First-principles computations of YxGa1–xAs-ternary alloys: a study on structural, electronic, optical and elastic properties. Bull. Mater. Sci. (2020). https://doi.org/10.1007/s12034-019-1978-y
Tran, F., Blaha, P.: Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. (2009). https://doi.org/10.1103/PhysRevLett.102.226401
Vurgaftman, I., Meyer, J.R., Ram-Mohan, L.R.: Band parameters for III-V compound semiconductors and their alloys. J. Appl. Phys. 89(11), 5815–5875 (2001)
Wang, F., Landau, D.P.: Efficient, multiple-range random walk algorithm to calculate the density of states. Phys. Rev. Lett. 86(10), 2050–2053 (2001)
Wang, X., Liu, W., Zhai, C., Yun, J., Zhang, Z.: DFT calculation on the electronic structure and optical properties of InxGa1−xN alloy semiconductors. Medziagotyra 26(2), 127–132 (2020)
Yousafa, M., Dalhatu, S.A., Murtaza, G., Khenata, R., Sajjad, M., Musa, A., Aliabad., Saeed, M.A.: Optoelectronic properties of XIn2S4 (X = Cd, Mg) thiospinels through highly accurate all-electron FP-LAPW method coupled with modified approximations. J. Alloys Compd. 625, 182–187 (2015)
Yousafa, M., Dalhatu, S.A., Murtaza, G., Khenata, R., Sajjad, M., Musa, A., Aliabad, R., Saeed, M.A., Zafara, M., Masood, M.K., Rizwan, M., Ziab, A., Ahmad, S., Akram, A., Baod, C., Shakil, M.: Theoretical study of structural, electronic, optical and elastic properties of AlxGa1–xP. Optik (stuttg) 182, 1176–1185 (2019)
Yu, R., Zhang, X.F.: Family of noble metal nitrides: first principles calculations of the elastic stability. Phys. Rev. B - Condens. Matter Mater. Phys. 72(5), 2–5 (2005)
Zhang, Z., Chai, C., Song, Y., Kong, L., Yang, Y.: A DFT study on physical properties of III-V compounds (AlN, GaN, AlP, and GaP) in the P3121 phase. Mater. Res. Express 8(2), 25908 (2021)
Acknowledgements
The authors would like to thank the University of Engineering and Technology Lahore Pakistan, International Islamic University Islamabad, University of Lahore Pakistan, University of Agriculture Faisalabad Pakistan, and Minhaj University Lahore Pakistan for computational facilities.
Funding
No funding was received for this research.
Author information
Authors and Affiliations
Contributions
SN, AWA, MA, NUH, RW, MT, MM, ZW and AA help to perform the material search, computational simulations and facilitate the writing of this manuscript. MA and KN help to analyze data, Graphical work and remained involved in valuable discussions on the research work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Ethical approval
Not applicable.
Human and animal rights statement
No human and/or animal studies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nabi, S., Anwar, A.W., Ahmad, M. et al. First-principles calculations to investigate structural, electronic and optical properties of In-doped aluminium antimonide alloy for optoelectronic applications. Opt Quant Electron 55, 799 (2023). https://doi.org/10.1007/s11082-023-05091-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-023-05091-2