
Abstract
The identification of mutations in the leucine-rich repeat kinase 2 (LRRK2) gene as a cause of autosomal dominant Parkinson’s disease (PD) was a major step forward in the genetic dissection of this disorder. However, what makes LRRK2 unique among the known PD-causing genes is that a low-penetrance mutation, Gly2019Ser, is a frequent determinant not only of familial, but also of sporadic PD in several populations from South Europe, North Africa and Middle East. Moreover, a different polymorphic variant, Gly2385Arg, is a frequent risk factor for PD among Chinese and Japanese populations. Currently, the Gly2019Ser and Gly2385Arg variants represent the most relevant PD-causing mutation and risk allele, respectively, linking the etiology of the familial and the sporadic forms of this disease. Understanding how the dysfunction of LRRK2 protein leads to neurodegeneration might provide crucial insights for unraveling the molecular mechanisms of PD and for developing disease-modifying therapies.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder, and the second most common neurodegenerative disease after Alzheimer’s disease (AD), with a prevalence of more than 1% after the age of 65 years [1]. The incidence of PD increases with age, and the number of patients is expected to double by the year 2030, due to aging of the population, improved diagnosis and prolonged survival, particularly in the developing countries [2].
PD is clinically defined by adult-onset, progressive parkinsonism (the combination of akinesia, resting tremor, and muscular rigidity), which displays a beneficial response to dopamine-replacement therapy [3]. In most patients, this clinical syndrome correlates with neuronal loss and gliosis in the substantia nigra pars compacta and other brain areas, and with formation of cytoplasmic inclusions called Lewy bodies (LB) and Lewy neurites in the surviving neurons.
The molecular mechanisms of PD remain mostly unknown. Several lines of evidence, including biochemical analysis, genomic and proteomic profiling of brain tissue, cell and animal models, implicated mitochondrial defects, oxidative stress, protein misfolding, proteasomal and lysosomal abnormalities in the pathogenesis [4–12]. However, there are many reciprocal interactions between these pathways, making it difficult to disentangle the primary and the secondary events. Moreover, the determinants of the preferential vulnerability of the dopaminergic system observed in PD remain unknown.
In most patients PD appears as a sporadic disorder. In 10–15% of cases the disease runs in families, but a Mendelian inheritance is rarely evident from the pedigree analysis. Yet, the ongoing identification of primary genetic defects in patients with inherited forms of PD is rapidly expanding the possible approaches to unravel the disease pathogenesis [13].
Five genes are today considered as definitely implicated in the etiology of PD. Mutations in the α-synuclein [14, 15] and leucine-rich repeat kinase 2 (LRRK2) [16, 17] gene cause autosomal dominant forms whereas mutations in the parkin [18], DJ-1 [19] and PINK1 (20) gene cause autosomal recessive forms of PD. LBs are found in the brain of patients with α-synuclein mutations and in most of the cases with LRRK2 mutations. On the other hand, LBs are not present in most of the patients with parkin mutations, while their occurrence in cases with DJ-1 or PINK1 mutations remains unknown.
The discovery that duplication and triplication of the whole α-synuclein gene is also a cause of autosomal dominant PD and of the related condition, dementia with LBs, links directly the over-expression of wild-type α-synuclein protein to the disease pathogenesis [15, 21, 22]. Moreover, common allelic variation in the α-synuclein gene might increase the risk for the sporadic form of PD [23]. A central role of α-synuclein in the pathogenesis of PD is further supported by the fact that wild type α-synuclein protein is the major component of the LBs and of other neuronal and glial inclusions found in PD, dementia with LBs, and multiple system atrophy, now collectively termed “synucleinopathies” [24, 25].
LRRK2 mutations as a cause of PD
A genome-wide search for linkage in a large Japanese pedigree with autosomal dominant, late-onset parkinsonism yielded the identification of a novel locus (PARK8) to the peri-centromeric region of chromosome 12 [26]. Interestingly, autopsy study of four affected members of this family revealed no LBs in the brain, a finding considered incompatible with a formal pathological diagnosis of PD. However, linkage to the same chromosomal region was later confirmed in two large families of European ancestry, segregating parkinsonism associated with different brain pathologies with or without LBs in different patients. This suggested PARK8 to be an important locus with a pleomorphic pathology [27].
Using positional cloning strategies, in the year 2004, LRRK2 was identified as the gene defective at the PARK8 locus [16, 17]. Soon thereafter, different groups identified a single LRRK2 mutation (c.G6055A) leading to a Gly2019Ser substitution in the encoded protein, which was present in familial and sporadic PD with unprecedented high frequency. A different mutation affecting the subsequent amino acid, Ile2020Thr, was detected as the cause of disease in the original Japanese PARK8 family [28]. The following two years have seen an explosion of research into the LRRK2 gene in PD and related disorders. Due to the large size of its open reading frame (more than 7.5 kb across 51 exons), a comprehensive screening of the entire LRRK2 coding region has been rarely performed so far, while in most studies large series of patients were screened only for one or few known mutations.
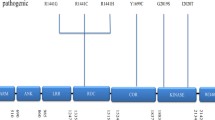
The screening of the complete coding region of LRRK2 revealed mutations in ∼10% of the PD cases with a family history compatible with autosomal dominant inheritance [29–32]; this figure by itself nominates LRRK2 as the most common known genetic cause of the disease. However, a note of caution is due here. For several mutations, co-segregation studies have been very limited or lacking; large series of ethnically matched controls have not always been tested; and assays are not available yet to study the effects of a given mutation on the function of the LRRK2 protein. Due to all these reasons, the pathogenic nature remains unclear for several of the LRRK2 mutations detected in PD cases, and at least some of these (for example: Arg1514Gln) might represent rare, benign variants [33, 34]. The uncertainties regarding the pathogenic nature of the mutations found in the LRRK2 (as well as in any other genes) are an important issue, which complicates the transfer of the results of the genetic screening into the clinics for diagnostic and genetic counseling purposes. To date, only five LRRK2 mutations (Arg1441Cys, Arg1441Gly, Tyr1699Cys, Gly2019Ser, and Ile2020Thr) can be considered as definitely disease causing, on the basis of a clear co-segregation with PD in large pedigrees and absence in large series of controls (Fig. 1).

Schematic representation of the human LRRK2 protein. The known functional domains are shown, the catalytic domains in black and the protein-protein interaction domains in grey. Five definitely PD-causing mutations and one PD-associated polymorphism are also displayed
Another very important aspect is the wide pathological spectrum associated with LRRK2 mutations (∼30 cases came to autopsy so far) [35]. Dopaminergic neuronal cell loss and gliosis in the substantia nigra are the common features in the patients carrying LRRK2 mutations [17]. In addition, classical LBs were found in the majority of cases, but in few, there was absence of inclusions, or only tau-positive or ubiquitin-positive inclusions were detected [31, 36–42]. These observations are based on a limited number of brains, and further studies are warranted. However, the pathologic pleomorphism seems a common theme for the different LRRK2 mutations, at least for Arg1441Cys, Tyr1699Cys, and Gly2019Ser, suggesting that our pathological definition of PD and related diseases has to be revised.
The Gly2019Ser story
Gly2019Ser is particularly important among the PD-causing mutations in LRRK2. This mutation was identified by several groups as a common cause of the disease, being detected initially in ∼5–6% of large cohorts of familial PD in Europe and US, and in ∼1–2% of sporadic PD from UK [43–47]. It is now clear that the frequency of Gly2019Ser in PD varies greatly across populations [48]. The results of the different studies are not easily comparable because of the different sample size, different methods for patient ascertainment, different definitions of “familial” versus “sporadic” disease, and different genotyping techniques, and much more work remains therefore ahead. The Gly2019Ser mutation has not been identified in three large series of Chinese patients [49–51], though it was rarely found in Indian [52] and Japanese patients [53, 54]. Studies in large referral series from the US population estimated a mutation frequency of up to ∼3% in familial and ∼0.7% in sporadic cases, respectively [55, 56]. This mutation seems present at lower frequency in patients from Northern Europe [57–61], than in those from Southern Europe such as Italy (∼5% of familial and ∼1% of sporadic cases) [62, 63], Spain and Portugal (up to ∼6–18% of familial and ∼3–6% of sporadic cases) [64–67](Ferreira et al. unpublished data). However, an extremely high prevalence is found among Arab patients from North Africa (∼37% of familial and ∼41% of sporadic cases) and among Ashkenazi Jewish patients (∼29% of familial and ∼13% of sporadic cases) [68, 69]. The prevalence of this mutation remains to be investigated in other large populations, such as those from Brazil, and other countries of Latin America.
Gly2019Ser represents clearly the first common pathogenic mutation identified in PD, establishing for the first time the proof-of-principle for a genetic determinant frequently involved in the classical, late-onset, sporadic forms of this disease.
Most of the patients carrying this mutation and living in disparate countries in Europe and America, share a common, very old founder haplotype [46, 62, 70], which likely originated from North Africa or Middle East ∼2,000 years ago or earlier [71]. A second haplotype has been detected in a few patients of European ancestry [71], while a third haplotype was found in Japanese patients [54]. The occurrence of Gly2019Ser in at least three different haplotypes suggests either an extremely old founder, or a mutational hot spot. Another hot spot might be represented by LRRK2 codon 1441, where three different mutations are known to occur (Arg1441Cys, Arg1441Gly, Arg1441His) [17, 29, 32, 72–74].
Mapping and cloning of genes for dominantly inherited diseases often relies on families with an exceptionally high number of affected individuals. This leads to an inherent ascertainment bias, and an overestimation of the mutation penetrance. However, after a causative mutation is identified, more accurate values of penetrance can be estimated in unselected, consecutive series of patients, ideally from population-based studies. This approach might yield considerably lower figures of penetrance. In the case of PARK8, a reduced penetrance of the underlying mutation was already suggested in the initial linkage study [26], and confirmed after the identification of the LRRK2 gene. Recent estimates of the lifetime penetrance of the Gly2019Ser mutation in large, hospital-based but otherwise unselected series of PD patients (US Jewish, US non-Jewish, and Italians) yielded values of ∼24–33% [56, 69, 75]. Yet, the penetrance might be different in other populations and additional studies are therefore warranted before Gly2019Ser testing is used for genetic counseling. Such a low penetrance explains the high Gly2019Ser prevalence among patients with sporadic PD, and its rare occurrence in controls (∼1%), particularly among the populations with the highest mutation frequencies such as Arabs and Ashkenazi Jews [68, 69].
Due to a lower frequency, the penetrance of other LRRK2 mutations is more difficult to estimate accurately, but reduced values are also suggested by analysis of pedigrees segregating the second most recurrent LRRK2 mutation, Arg1441Cys [62, 76].
The clinical phenotype of Gly2019Ser-positive patients appears very similar, or indistinguishable from that of the classical form of PD, but a wide range of onset age is evident [45, 55, 56, 62, 63, 77]. Several patients, mostly from Tunisia and Algeria, were identified who carry the Gly2019Ser mutation in homozygous state [78–80]. This is likely due to the high prevalence of the mutation, and the high frequency of consanguineous marriages in those populations. Homozygous carriers of this mutation seem not to develop PD at an earlier age, nor to have a more severe disease, or a more aggressive course, compared to the heterozygous carriers [80]. However, it is difficult to draw firm conclusions, as the clinical spectrum associated in heterozygous mutation carriers is also very broad. Interestingly, the penetrance might be higher in homozygous carriers [79], arguing for the presence of a mutation dosage effect. The low penetrance and variable phenotypic expressivity of the mutation suggest the existence of further important modifiers, which might include other genetic as well as non-genetic factors. Their identification is an important area of the current research.
Gly2385Arg: a common risk allele for PD in Asia
At the beginning of the year 2006, we found that a different LRRK2 variant, Gly2385Arg, is a polymorphism in the Han Chinese population from Taiwan (frequency of heterozygous carriers ∼5% among controls), and it is significantly more frequent (∼10%) among PD cases [81]. We therefore proposed that Gly2385Arg is a common risk factor for PD in the Han population. Interestingly, this variant was initially detected in a single, small PD family from Taiwan [29], but it has not been observed so far among whites [30–32], and it appears therefore specific for the Asian population. The association between Gly2385Arg and PD has now been confirmed in at least four independent replication studies, involving more than 2000 individuals (three targeting the Chinese, and one the Japanese population) [82–85] (Table 1).
Using the observed frequency of the Gly2385Arg genotype among controls and the observed value of odds ratio as estimates of the risk genotype frequency in the general population, and of the relative risk, respectively, one can calculate a population attributable risk of ∼4% for the Gly2385Arg heterozygous genotype in the Han Chinese population [82]. Cross-sectional case control studies are prone to several biases, including survival bias. It is therefore crucial to replicate this finding also in large prospective studies. However, the replication of the association in the same direction-of-effect and with similar effect size (odds ratio ∼2.5 in most studies) in four independent, large samples of different geographic and ethnic origin (Table 1), and the potential functional effects of this missense, non-conservative variant, all strongly support the contention that this represents a real, causal association. Gly2385Arg might be the first identified genetic risk factor for the common PD form in the Asian population, and the most frequent genetic determinant of PD worldwide, also considering the large and expanding size of the Chinese population (projected number of ∼5 millions patients by the year 2030) [2].
As observed for the Gly2019Ser carriers, the clinical spectrum in PD patients who carry the Gly2385Arg variant is very broad and indistinguishable from that of the cases who do not carry it.
The Gly2385Arg variant is located at the surface of the C-terminal WD40 domain of the LRRK2 protein, where it introduces an additional, net positive electric charge. WD40 domains are involved in protein–protein interactions, and they might be important for the formation of complexes between LRRK2 and other proteins, or for the LRRK2 dimerization. It is possible that the Gly2385Arg variant increases the risk of PD by affecting these biochemical properties of the LRRK2 protein.
The LRRK2 protein
LRRK2 mutation causes a disease that most closely resembles the common forms of PD. The LRRK2 protein is likely to be a very important player in the pathogenesis of PD in general, and the pharmacological manipulation of the LRRK2 activity might become a future important therapeutic strategy. It is therefore urgent to unravel the biology of the LRRK2 protein, and how its mutation leads to neurodegeneration, but very little is known about these crucial aspects.
LRRK2 encodes a 2,527 amino acids protein of unknown function, belonging to the “ROCO” group within the Ras/GTPase superfamily [86], and characterized by the presence of several conserved domains: a Roc (Ras in complex proteins) and a COR (C-terminal of Roc) domain, together with a leucine-rich repeat region, a WD40 domain, two ankyrin-like motifs, and a protein kinase catalytic domain (Fig. 1). Review of the ROCO family members reveals involvement in diverse cellular processes (regulation of cell polarity, chemotaxis, cytokinesis, cytoskeletal rearrangements, and programmed cell death), making impossible to predict the function of human LRRK2 on the basis of homology (reviewed in [86, 87]).
Initial studies suggest that the LRRK2 mRNA [88–91] and the LRRK2 protein [39, 88, 92, 93] are broadly expressed throughout the brain, including nigral neurons, and that the LRRK2 protein shows a cytosolic localization, perhaps in association with membranous structures [93]. There is also evidence that the LRRK2 protein regulates the length and branching of neurites and this function might be impaired by PD-causing mutations [94].
LRRK2 immunoreactivity has been reported in some LBs from PD brains [92, 95]. However, most of the currently available LRRK2 antibodies lack optimal sensitivity and specificity, and further investigations are definitely warranted.
One of the two predicted catalytic domains (GTPase and kinase) represents likely the output activity of LRRK2. Small GTPases (Rho, Rac, Cdc42) usually act upstream of protein kinases. By analogy, the GTPase domain might regulate the LRRK2 kinase domain via intramolecular signaling. Whether the LRRK2 kinase activity is required for the phosphorylation of target proteins, or whether it plays an auto-regulatory role, is currently unclear. The PD-causing mutations replace highly conserved residues, but, in addition, the Glycine2019 residue is remarkable because it is conserved in all human kinase domains. These mutations could destabilize the kinase domain, resulting in loss-of-function of the kinase activity, and suggesting haploinsufficiency as disease mechanism. Another possibility is that Gly2019Ser and other mutations enhance the kinase activity. Of note, the three known PD-causing mutations in the kinase domain (Ile2012Thr, Gly2019Ser, and Ile2020Thr) all introduce novel potential auto-phosphorylation sites, and similar mutations in the activation segment of other kinases induce hyper-activity [96]. This mechanism would confer a gain of function to the mutant protein, fitting with the dominant pattern of inheritance seen in families with LRRK2 mutations.
Over-expressing the human wild-type LRRK2 protein in different cell systems is associated with formation of cytoplasmic inclusions [92, 97]. Moreover, LRRK2 shows protein kinase activity in vitro toward generic substrates and is capable of auto-phosphorylation [92, 98, 99]. Importantly, some of the PD-causing mutations (particularly those located in the kinase domain) appear to enhance the LRRK2 kinase activity in vitro, as well as the inclusion formation, and they induce cell toxicity and ultimately, cell death [92, 97–99]. LRRK2 also displays GTP-binding properties in vitro, and GTP binding seems required for the kinase domain of LRRK2 to be in an active state [100–102]. However, LRRK2 seems devoid of intrinsic GTPase activity, suggesting the involvement of other interacting proteins, such as GTPase activating proteins (GAPs), and guanine nucleotide exchange factors (GEFs) [100–102]. Here, the caveat is that all these findings need validation using in vivo models, and after the (currently unknown) physiological interactors and substrates of the LRRK2 protein are identified. A study focusing on the homologue LRRK1 protein came to the opposite conclusion that the LRRK1 kinase activity might be decreased by amino acid substitutions corresponding to the PD-causing mutations in LRRK2 [103]. Much more work remains ahead in order to understand the biology and pathology of this complex, fascinating protein.
Conclusions
The discovery of LRRK2 mutations in PD led to a turning point in the field. For the first time, gene mutations, and particularly the low-penetrance Gly2019Ser mutation prove to be a frequent genetic determinant of familial and sporadic forms of this disease in several populations; the Gly2385Arg polymorphic variant is a common risk factor for PD in Asia. In both cases, the associated clinical phenotype is indistinguishable from the classical, late-onset PD, and brain study reveals a broad pathological spectrum, which includes in most cases the typical LB pathology. Importantly, this frequent low-penetrance mutation and the frequent risk allele provide etiological links between the familial and the sporadic forms of PD.
Elucidating the function of the LRRK2 protein and how LRRK2 dysfunction leads to neurodegeneration might provide crucial insights for understanding the molecular mechanisms of PD and yield novel avenues to the development of a cure. The pharmacological modulation of one or both catalytic LRRK2 activities could become innovative, important therapeutic strategy for all patients with PD and related neurodegenerative diseases.
References
de Lau LM, Breteler MM (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5:525–535
Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68:384–386
Fahn S (2003) Description of Parkinson’s disease as a clinical syndrome. Ann NY Acad Sci 991:1–14
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3:1301–1306
McNaught KS, Perl DP, Brownell AL, Olanow CW (2004) Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann Neurol 56:149–162
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305:1292–1295
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39:889–909
Cookson MR (2005) The biochemistry of Parkinson’s disease. Annu Rev Biochem 74:29–52
Moore DJ, West AB, Dawson VL, Dawson TM (2005) Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28:57–87
Grunblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, Youdim MB (2004) Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm 111:1543–1573
Moran LB, Duke DC, Deprez M, Dexter DT, Pearce RK, Graeber MB (2006) Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson’s disease. Neurogenetics 7:1–11
Jin J, Hulette C, Wang Y, Zhang T, Pan C, Wadhwa R, Zhang J (2006) Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol Cell Proteomics 5:1193–1204
Bonifati V, Oostra BA, Heutink P (2004) Unraveling the pathogenesis of Parkinson’s disease—the contribution of monogenic forms. Cell Mol Life Sci 61:1729–1750
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) Alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, de Munain AL, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44:595–600
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160
Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364:1169–1171
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364:1167–1169
Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, Berg D, Wullner U, Meitinger T, Gasser T (2005) Multiple regions of alpha-synuclein are associated with Parkinson’s disease. Ann Neurol 57:535–541
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840
Spillantini MG, Goedert M (2000) The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann NY Acad Sci 920:16–27
Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F (2002) A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 51:296–301
Zimprich A, Muller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M, Lockhart P, Strongosky A, Kachergus J, Calne DB, Stoessl J, Uitti RJ, Pfeiffer RF, Trenkwalder C, Homann N, Ott E, Wenzel K, Asmus F, Hardy J, Wszolek Z, Gasser T (2004) The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. Am J Hum Genet 74:11–19
Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, Tsuji S, Obata F (2005) An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol 57:918–921
Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T, Hulihan MM, Cobb SA, Wu RM, Lu CS, Lahoz C, Wszolek ZK, Farrer MJ (2005) Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics 6:171–177
Berg D, Schweitzer K, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T, Wszolek ZK, Gasser T (2005) Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease. Brain 128:3000–3011
Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R, Gayton J, Lennox G, Revesz T, Nicholl D, Bhatia KP, Quinn N, Brooks D, Lees AJ, Davis MB, Piccini P, Singleton AB, Wood NW (2005) Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128:2786–2796
Di Fonzo A, Tassorelli C, De Mari M, Chien HF, Ferreira J, Rohe CF, Riboldazzi G, Antonini A, Albani G, Mauro A, Marconi R, Abbruzzese G, Lopiano L, Fincati E, Guidi M, Marini P, Stocchi F, Onofrj M, Toni V, Tinazzi M, Fabbrini G, Lamberti P, Vanacore N, Meco G, Leitner P, Uitti RJ, Wszolek ZK, Gasser T, Simons EJ, Breedveld GJ, Goldwurm S, Pezzoli G, Sampaio C, Barbosa E, Martignoni E, Oostra BA, Bonifati V (2006) Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson’s disease. Eur J Hum Genet 14:322–331
Nichols WC, Marek DK, Pauciulo MW, Pankratz N, Halter CA, Rudolph A, Shults CW, Wojcieszek J, Foroud T (2006) R1514Q substitution in Lrrk2 is not a pathogenic Parkinson’s disease mutation. Mov Disord Epub ahead of print, Dec 5, DOI 10.1002/mds.21233
Toft M, Mata IF, Ross OA, Kachergus J, Hulihan MM, Haugarvoll K, Stone JT, Blazquez M, Gibson JM, Aasly JO, White LR, Lynch T, Adler CH, Gwinn-Hardy K, Farrer MJ (2007) Pathogenicity of the LRRK2 R1514Q substitution in Parkinson’s disease. Mov Disord Epub ahead of print, Jan 10, DOI 10.1002/mds.21217
Bonifati V (2006) The Pleomorphic pathology of inherited Parkinson’s disease:lessons from LRRK2. Curr Neurol Neurosci Rep 6:355–357
Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, Strongosky AJ, Zimprich A, Muller-Myhsok B, Farrer MJ, Gasser T, Calne DB, Dickson DW (2004) Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 62:1619–1622
Wszolek ZK, Vieregge P, Uitti RJ, Gasser T, Yasuhara O, Mc Geer P, Berry K, Calne DB, Vingerhoets FJG, Klein C, Pfeiffer RF (1997) German-Canadian family (Family A) with parkinsonism, amyotrophy, and dementia—longitudinal observations. Parkinsonism Relat Disord 3:125–139
Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, Litvan I, Gordon MF, Wszolek ZK, Farrer MJ, Dickson DW (2006) Lrrk2 and Lewy body disease. Ann Neurol 59:388–393
Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM (2006) Biochemical and pathological characterization of Lrrk2. Ann Neurol 59:315–322
Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ (2006) Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 67:1506–1508
Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E (2007) G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. J Neurol Neurosurg Psychiatry Published Online First: 8 January 2007. doi:10.1136/jnnp.2006.107904
Giordana MT, D’Agostino C, Albani G, Mauro A, Di Fonzo A, Antonini A, Bonifati V (2007) Neuropathology of Parkinson’s disease associated with the LRRK2 Ile1371Val mutation. Mov Disord 22:275–278
Di Fonzo A, Rohe CF, Ferreira J, Chien HF, Vacca L, Stocchi F, Guedes L, Fabrizio E, Manfredi M, Vanacore N, Goldwurm S, Breedveld G, Sampaio C, Meco G, Barbosa E, Oostra BA, Bonifati V (2005) A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365:412–415
Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T, Wood NW (2005) A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 365:415–416
Nichols WC, Pankratz N, Hernandez D, Paisan-Ruiz C, Jain S, Halter CA, Michaels VE, Reed T, Rudolph A, Shults CW, Singleton A, Foroud T (2005) Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 365:410–412
Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ, Toft M (2005) Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 76:672–680
Hernandez DG, Paisan-Ruiz C, McInerney-Leo A, Jain S, Meyer-Lindenberg A, Evans EW, Berman KF, Johnson J, Auburger G, Schaffer AA, Lopez GJ, Nussbaum RL, Singleton AB (2005) Clinical and positron emission tomography of Parkinson’s disease caused by LRRK2. Ann Neurol 57:453–456
Bonifati V (2006) The LRRK2-G2019S mutation: opening a novel era in Parkinson’s disease genetics. Eur J Hum Genet 14:1061–1062
Lu CS, Simons EJ, Wu-Chou YH, Di Fonzo A, Chang HC, Chen RS, Weng YH, Rohe CF, Breedveld GJ, Hattori N, Gasser T, Oostra BA, Bonifati V (2005) The LRRK2 I2012T, G2019S, and I2020T mutations are rare in Taiwanese patients with sporadic Parkinson’s disease. Parkinsonism Relat Disord 11:521–522
Tan EK, Shen H, Tan LC, Farrer M, Yew K, Chua E, Jamora RD, Puvan K, Puong KY, Zhao Y, Pavanni R, Wong MC, Yih Y, Skipper L, Liu JJ (2005) The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson’s disease patients. Neurosci Lett 384:327–329
Fung HC, Chen CM, Hardy J, Hernandez D, Singleton A, Wu YR (2006) Lack of G2019S LRRK2 mutation in a cohort of Taiwanese with sporadic Parkinson’s disease. Mov Disord 21:880–881
Punia S, Behari M, Govindappa ST, Swaminath PV, Jayaram S, Goyal V, Muthane UB, Juyal RC, Thelma BK (2006) Absence/rarity of commonly reported LRRK2 mutations in Indian Parkinson’s disease patients. Neurosci Lett 409:83–88
Tomiyama H, Li Y, Funayama M, Hasegawa K, Yoshino H, Kubo S, Sato K, Hattori T, Lu CS, Inzelberg R, Djaldetti R, Melamed E, Amouri R, Gouider-Khouja N, Hentati F, Hatano Y, Wang M, Imamichi Y, Mizoguchi K, Miyajima H, Obata F, Toda T, Farrer MJ, Mizuno Y, Hattori N (2006) Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson’s disease patients from 18 countries. Mov Disord 21:1102–1108
Zabetian CP, Morino H, Ujike H, Yamamoto M, Oda M, Maruyama H, Izumi Y, Kaji R, Griffith A, Leis BC, Roberts JW, Yearout D, Samii A, Kawakami H (2006) Identification and haplotype analysis of LRRK2 G2019S in Japanese patients with Parkinson disease. Neurology 67:697–699
Kay DM, Zabetian CP, Factor SA, Nutt JG, Samii A, Griffith A, Bird TD, Kramer P, Higgins DS, Payami H (2006) Parkinson’s disease and LRRK2: frequency of a common mutation in US movement disorder clinics. Mov Disord 21:519–523
Clark LN, Wang Y, Karlins E, Saito L, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K (2006) Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology 67:1786–1791
Aasly JO, Toft M, Fernandez-Mata I, Kachergus J, Hulihan M, White LR, Farrer M (2005) Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann Neurol 57:762–765
Carmine Belin A, Westerlund M, Sydow O, Lundstromer K, Hakansson A, Nissbrandt H, Olson L, Galter D (2006) Leucine-rich repeat kinase 2 (LRRK2) mutations in a Swedish Parkinson cohort and a healthy nonagenarian. Mov Disord 21:1731–1734
Williams-Gray CH, Goris A, Foltynie T, Brown J, Maranian M, Walton A, Compston DA, Sawcer SJ, Barker RA (2006) Prevalence of the LRRK2 G2019S mutation in a UK community based idiopathic Parkinson’s disease cohort. J Neurol Neurosurg Psychiatry 77:665–667
Bialecka M, Hui S, Klodowska-Duda G, Opala G, Tan EK, Drozdzik M (2005) Analysis of LRRK2 G2019S and I2020T mutations in Parkinson’s disease. Neurosci Lett 390:1–3
Pchelina SN, Yakimovskii AF, Ivanova ON, Emelianov AK, Zakharchuk AH, Schwarzman AL (2006) G2019S LRRK2 mutation in familial and sporadic Parkinson’s disease in Russia. Mov Disord 21:2234–2236
Goldwurm S, Di Fonzo A, Simons EJ, Rohe CF, Zini M, Canesi M, Tesei S, Zecchinelli A, Antonini A, Mariani C, Meucci N, Sacilotto G, Sironi F, Salani G, Ferreira J, Chien HF, Fabrizio E, Vanacore N, Dalla Libera A, Stocchi F, Diroma C, Lamberti P, Sampaio C, Meco G, Barbosa E, Bertoli-Avella AM, Breedveld GJ, Oostra BA, Pezzoli G, Bonifati V (2005) The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J Med Genet 42:e65
Goldwurm S, Zini M, Di Fonzo A, De Gaspari D, Siri C, Simons EJ, van Doeselaar M, Tesei S, Antonini A, Canesi M, Zecchinelli A, Mariani C, Meucci N, Sacilotto G, Cilia R, Isaias IU, Bonetti A, Sironi F, Ricca S, Oostra BA, Bonifati V, Pezzoli G (2006) LRRK2 G2019S mutation and Parkinson’s disease: a clinical, neuropsychological and neuropsychiatric study in a large Italian sample. Parkinsonism Relat Disord 12:410–419
Gaig C, Ezquerra M, Marti MJ, Munoz E, Valldeoriola F, Tolosa E (2006) LRRK2 mutations in Spanish patients with Parkinson disease: frequency, clinical features, and incomplete penetrance. Arch Neurol 63:377–382
Infante J, Rodriguez E, Combarros O, Mateo I, Fontalba A, Pascual J, Oterino A, Polo JM, Leno C, Berciano J (2006) LRRK2 G2019S is a common mutation in Spanish patients with late-onset Parkinson’s disease. Neurosci Lett 395:224–226
Mata IF, Ross OA, Kachergus J, Huerta C, Ribacoba R, Moris G, Blazquez M, Guisasola LM, Salvador C, Martinez C, Farrer M, Alvarez V (2006) LRRK2 mutations are a common cause of Parkinson’s disease in Spain. Eur J Neurol 13:391–394
Bras JM, Guerreiro RJ, Ribeiro MH, Januario C, Morgadinho A, Oliveira CR, Cunha L, Hardy J, Singleton A (2005) G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov Disord 20:1653–1655
Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A (2006) LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med 354:422–423
Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, Lipton RB, Soto-Valencia J, Risch N, Bressman SB (2006) LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 354:424–425
Lesage S, Leutenegger A-L, Ibanez P, Janin S, Lohmann E, Durr A, Brice A (2005) LRRK2 haplotype analyses in European and North African families with Parkinson’s disease: a common founder for the G2019S mutation dating from the 13th century. Am J Hum Genet 77:330–332
Zabetian CP, Hutter CM, Yearout D, Lopez AN, Factor SA, Griffith A, Leis BC, Bird TD, Nutt JG, Higgins DS, Roberts JW, Kay DM, Edwards KL, Samii A, Payami H (2006) LRRK2 G2019S in families with Parkinson disease who originated from Europe and the Middle East: evidence of two distinct founding events beginning two millennia ago. Am J Hum Genet 79:752–758
Simon-Sanchez J, Marti-Masso JF, Sanchez-Mut JV, Paisan-Ruiz C, Martinez-Gil A, Ruiz-Martinez J, Saenz A, Singleton AB, Lopez de Munain A, Perez-Tur J (2006) Parkinson’s disease due to the R1441G mutation in Dardarin: a founder effect in the Basques. Mov Disord 21:1954–1959
Zabetian CP, Samii A, Mosley AD, Roberts JW, Leis BC, Yearout D, Raskind WH, Griffith A (2005) A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology 65:741–744
Spanaki C, Latsoudis H, Plaitakis A (2006) LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology 67:1518–1519
Goldwurm S, Zini M, Mariani L, Tesei S, Miceli R, Sironi F, Clementi M, Bonifati V, Pezzoli G (2007) Evaluation of LRRK2 G2019S penetrance: relevance for genetic counseling in Parkinson disease. Neurology E-pub ahead of print, January 10, 2007, doi:10.1212/01.wnl.0000254483.19854.ef
Hedrich K, Winkler S, Hagenah J, Kabakci K, Kasten M, Schwinger E, Volkmann J, Pramstaller PP, Kostic V, Vieregge P, Klein C (2006) Recurrent LRRK2 (Park8) mutations in early-onset Parkinson’s disease. Mov Disord 21:1506–1510
Isaias IU, Benti R, Goldwurm S, Zini M, Cilia R, Gerundini P, Di Fonzo A, Bonifati V, Pezzoli G, Antonini A (2006) Striatal dopamine transporter binding in Parkinson’s disease associated with the LRRK2 Gly2019Ser mutation. Mov Disord 21:1144–1147
Lesage S, Ibanez P, Lohmann E, Pollak P, Tison F, Tazir M, Leutenegger AL, Guimaraes J, Bonnet AM, Agid Y, Durr A, Brice A (2005) G2019S LRRK2 mutation in French and North African families with Parkinson’s disease. Ann Neurol 58:784–787
Ishihara L, Gibson RA, Warren L, Amouri R, Lyons K, Wielinski C, Hunter C, Swartz JE, Elango R, Akkari PA, Leppert D, Surh L, Reeves KH, Thomas S, Ragone L, Hattori N, Pahwa R, Jankovic J, Nance M, Freeman A, Gouider-Khouja N, Kefi M, Zouari M, Ben Sassi S, Ben Yahmed S, El Euch-Fayeche G, Middleton L, Burn DJ, Watts RL, Hentati F (2006) Screening for Lrrk2 G2019S and clinical comparison of Tunisian and North American Caucasian Parkinson’s disease families. Mov Disord 22:55–61
Ishihara L, Warren L, Gibson R, Amouri R, Lesage S, Durr A, Tazir M, Wszolek ZK, Uitti RJ, Nichols WC, Griffith A, Hattori N, Leppert D, Watts R, Zabetian CP, Foroud TM, Farrer MJ, Brice A, Middleton L, Hentati F (2006) Clinical features of Parkinson disease patients with homozygous leucine-rich repeat kinase 2 G2019S mutations. Arch Neurol 63:1250–1254
Di Fonzo A, Wu-Chou YH, Lu CS, van Doeselaar M, Simons EJ, Rohe CF, Chang HC, Chen RS, Weng YH, Vanacore N, Breedveld GJ, Oostra BA, Bonifati V (2006) A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson’s disease risk in Taiwan. Neurogenetics 7:133–138
Tan EK, Zhao Y, Skipper L, Tan MG, Di Fonzo A, Sun L, Fook-Chong S, Tang S, Chua E, Yuen Y, Tan L, Pavanni R, Wong MC, Kolatkar P, Lu CS, Bonifati V, Liu JJ (2007) The LRRK2 Gly2385Arg variant is associated with Parkinson’s disease: genetic and functional evidence. Hum Genet 120:857–863
Fung HC, Chen CM, Hardy J, Singleton AB, Wu YR (2006) A common genetic factor for Parkinson disease in ethnic Chinese population in Taiwan. BMC Neurol 6:47
Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K, Ross OA, Wu RM (2007) Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord Epub ahead of print, Jan 9, doi:10.1016/j.parkreldis.2006.12.001
Funayama M, Li Y, Yoshino H, Imamich Y, Tomiyama H, Yamamoto M, Murata M, Toda T, Hattori N, Mizuno Y (2006) A variation on LRRK2 is associated with Parkinson’s disease in Asian population. Mov Disord 21(Suppl. 15):S414
Bosgraaf L, Van Haastert PJ (2003) Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta 1643:5–10
Marin I (2006) The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol 23:2423–2433
Taymans JM, Van den Haute C, Baekelandt V (2006) Distribution of PINK1 and LRRK2 in rat and mouse brain. J Neurochem 98:951–961
Melrose H, Lincoln S, Tyndall G, Dickson D, Farrer M (2006) Anatomical localization of leucine-rich repeat kinase 2 in mouse brain. Neuroscience 139:791–794
Simon-Sanchez J, Herranz-Perez V, Olucha-Bordonau F, Perez-Tur J (2006) LRRK2 is expressed in areas affected by Parkinson’s disease in the adult mouse brain. Eur J Neurosci 23:659–666
Galter D, Westerlund M, Carmine A, Lindqvist E, Sydow O, Olson L (2006) LRRK2 expression linked to dopamine-innervated areas. Ann Neurol 59:714–719
Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 23:329–341
Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL (2006) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol 60:557–569
MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A (2006) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52:587–593
Zhu X, Babar A, Siedlak SL, Yang Q, Ito G, Iwatsubo T, Smith MA, Perry G, Chen SG (2006) LRRK2 in Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener 1:17
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954
Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA (2005) Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci USA 102:18676–18681
West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA 102:16842–16847
Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O’Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M (2006) The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet 15:223–232
Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9:1231–1233
West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM (2007) Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 16:223–232
Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, Iwatsubo T (2007) GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson’s disease. Biochemistry 46:1380–1388
Korr D, Toschi L, Donner P, Pohlenz HD, Kreft B, Weiss B (2006) LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal 18:910–920
Acknowledgments
I wish to thank the patients and family relatives for their contribution, and the many clinical collaborators involved in our studies on the genetics of PD. Dr. Alessio Di Fonzo and Dr. Dorothea Schweiger performed most of the LRRK2 genetic and protein studies in my laboratory. I thank Tom de Vries-Lentsch, Erasmus MC, Rotterdam, for artwork. Our studies are supported by grants from the Internationaal Parkinson Fonds (The Netherlands), the Hersenstichting Nederland, and the Erasmus MC, Rotterdam (Erasmus Fellowship 2006).
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue dedicated to Dr. Moussa Youdim.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bonifati, V. LRRK2 Low-penetrance Mutations (Gly2019Ser) and Risk Alleles (Gly2385Arg)—Linking Familial and Sporadic Parkinson’s Disease. Neurochem Res 32, 1700–1708 (2007). https://doi.org/10.1007/s11064-007-9324-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-007-9324-y