
Abstract
A green and efficient one-pot multi-component protocol was developed for the synthesis of some novel dihydrochromeno[4,3-b]pyrrol-3-yl derivatives through the reaction of arylglyoxals, malono derivatives, and different 4-amino coumarins in ethanol at reflux condition. In this method, all products were obtained in good to excellent yield. Next, all synthesized derivatives were evaluated for their α-glucosidase inhibitory activity. Most of the compounds displayed potent inhibitory activities with IC50 values in the range of 48.65 ± 0.01–733.83 ± 0.10 μM compared to the standard inhibitor acarbose (IC50 = 750.90 ± 0.14 μM). The kinetic study of compound 5e as the most potent derivative (IC50 = 48.65 ± 0.01 μM) showed a competitive mechanism with a Ki value of 42.6 µM. Moreover, docking studies revealed that dihydrochromeno[4,3-b]pyrrol-3-yl effectively interacted with important residues in the active site of α-glucosidase.
Similar content being viewed by others

Introduction
Multi-component reactions (MCRs) are convergent reactions in which three or more raw materials react together to form a product so that all or most of the atoms form the new product [1]. MCRs are of particular importance because of their advantages such as simplicity of operation, reduction of separation and treatment steps, minimization of energy, time, cost, and waste generation [2,3,4,5]. Therefore, many researchers in the field of pharmaceutical, biological, and organic chemistry have considered the design and use of MCRs.
Type 2 diabetes mellitus (T2DM) is recognized as one of the most extensive global health challenges in the modern world affecting approximately 462 million individuals corresponding to 6.28% of the world’s population with over 1 million deaths per year [6, 7]. Diabetes is also a risk factor for cardiovascular disease, cognitive decline, and Alzheimer's dementia [8]. The concerning trends in incidence, prevalence, and mortality of T2DM as well as its economical burden promote researchers to develop new agents to reduce exposure to high glucose levels in the postprandial state [9].
In this regard, α-glucosidase is an attractive target. α-glucosidase (EC.3.2.1.20) is an essential enzyme located at the brush border of the intestines which hydrolyses the 1,4-α-glycosidic linkages of oligosaccharides, trisaccharides, and disaccharides to monosaccharides [10]. As a result, the increased level of simple sugars can be absorbed into the blood from the intestine. With the help of the bloodstream, these simple sugars enter the cells and are converted into energy with the help of insulin. Without insulin (inadequate insulin secretion or insulin resistance), glucose stays in the bloodstream, keeping blood sugar levels high which leads to hyperglycemia, impaired glucose tolerance, and at late-stage type II diabetes [11,12,13].
α-glucosidase inhibitors are used to reduce the hydrolyze, digestion, and absorption of carbohydrates as well as suppress postprandial hyperglycemia [14]. In the last few years, various molecules have been reported as potential inhibitors of α-glucosidase with natural origin such as polyphenols, terpenoids, flavonoids, alkaloids, and saponins [15, 16]. Acarbose, as an FDA-approved α-glucosidase inhibitor, is administered orally to control blood sugar levels. Nojirimycin, miglitol, and voglibose are other market anti-α-glucosidase drugs for T2DM [17, 18]. Meanwhile, various synthetic small-molecules of α-glucosidase inhibitors have been developed bearing imidazole [19], pyrazoles [20], quinazolinone [21], isatin [22], xanthone [23], and azole [24] groups.
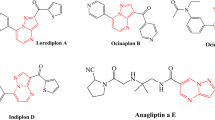
Coumarin-fused heterocycles are among the most interesting and widely used compounds because of their applications such as anti-tumour [25], antibacterial [26], antifungal [26], anticoagulant [27], anti-inflammatory [28], antiviral [28] and anti-HIV activities [29]. Among them, pyrrole-fused derivatives of coumarin are important classes of natural marine materials, and a number of them showed significant biological and medicinal properties (Fig. 1). Marine alkaloids Ningalin B and Lamellarin D with chromenopyrrole moiety in their structures demonstrate potential treatments in central nervous system disorders [30,31,32,33,34]. Also, Lamellarins D, K, and M are cytotoxic agents to a wide range of cancer cell lines. Recently, due to the biological importance of chromenopyrroles, the synthesis of this type of compounds has been considered in different research groups [34,35,36,37,38,39].

Biologically active coumarin-fused pyrrole derivatives
In continuation of our research on the synthesis of chromene and pyrrole [40,41,42,43], herein, a series of chromeno[4,3-b]pyrrol-3-yl were designed and synthesized through the MCRs. All derivatives were evaluated in vitro as possible anti-α-glucosidase agents, and the mode of inhibition was rationalized in silico through molecular docking studies.
Results and discussion
Design
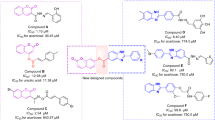
To design a novel and efficient series of α-glucosidase inhibitors, the structural feature of some potent inhibitors with the possibility of MCRs synthesis reported in the literature was evaluated. Compounds containing chromenone (coumarin) rings have become an emerging anti-diabetic scaffold, recently (Fig. 2). Compound A was an attractive derivative bearing coumarin moiety with an IC50 value of 2.7 μM. Interestingly, the replacement of the coumarin ring with other groups such as chloro, bromo, or nitro-phenyl showed a reduction in the inhibitory potency significantly [44]. In another study, coumarin containing thiazole-carbohydrates series demonstrated moderate to high potency with an IC50 value in the range of 6.2–81 μM. Structure–activity relationship (SAR) studies proposed that electron-withdrawing substituents at phenyl of carbohydrazone improve inhibitory activity significantly. Docking analysis of compound B represented compact conformation through hydrophobic interactions of Pro240, Phe157, and Phe177 residues with coumarin ring [45]. Another series of coumarin-thiazoles were synthesized with an IC50 value in the range of 0.12–16.20 μM as compared to standard acarbose (IC50 = 38.25 μM). Docking study of the most potent derivative (compound C) displayed interactions with Asn241, Arg312, and Phe300. Also, polar interactions were observed in coumarin’s oxygen and Asn241 residue [46].

Chemical structures of some reported α-glucosidase inhibitors and newly designed compound
On the other hands, pyrroles and their derivatives have attracted attention because of the ability of these compounds to act as glycosidase inhibitors so that pyrrole can mimic oxocarbocation intermediate structure, enable tight binding and strongly inhibit the enzyme [47]. Accordingly, there are some reports concerning α-glucosidase inhibitors possessing pyrrole rings. Compound D was reported to exhibit an IC50 of 0.08 μM against α-glucosidase [47]. Jadhaval et al. designed phenyl-1H-pyrrole derivatives and evaluated their α-glucosidase inhibitory activity. Compounds E showed excellent activity as compared to standard acarbose (IC50 = 0.3 μmol/mL). According to molecular docking study, the pyrrole ring of the molecule E occupied the cavity made by the residues, namely Trp58, Trp59, Glu63, Val163, and Leu165, while the phenyl ring occupied the pocket formed by the residues Hie101, Leu162, Arg195, Ala198, and Glu233 [48]. In another study, natural pyrrole-bearing compounds were found in G. frondosa for the first time. Compounds F and G showed potent inhibition against α-glucosidase compared with acarbose with an IC50 value of 642.99 μM [49].
According to our limited literature review, compounds bearing both chromenone and pyrrole in particular, as anti- α-glucosidase agents have not been proposed yet. As a result, in the current study, molecular hybridization approaches were applied to design these targeted compounds which are anticipated to possess potent α-glucosidase inhibitory activity. Subsequently, fourteen chromeno[4,3-b]pyrrol derivatives were synthesized through MCRs and were evaluated against α-glucosidase. Moreover, the kinetic and docking studies of the most potent compound were performed to better understand the mode of inhibition and interactions with the enzyme.
Synthesis
Arylglyoxal derivatives were prepared from the oxidation of aryl methyl ketones using selenium dioxide as oxidant (Scheme 1) [50].

Preparation of arylglyoxals from the oxidation of aryl methyl ketones using SeO2 as oxidant
Then, to obtain the best reaction conditions for the synthesis of chromeno-pyrrole derivatives, the reaction between phenylglyoxal 1a (1 mmol), malononitrile 2a (1 mmol), and 4-aminocoumarin 3a (1 mmol) was studied as a model reaction in different solvents and reaction temperatures. As can be seen in Table 1, the best yield and shorter reaction times were obtained when the reaction was carried out in ethanol at reflux conditions (Table 1, entry 7).
Subsequently, the efficiency of this method was explored under the optimized reaction conditions for the condensation of different arylglyoxals, 4-amino coumarins, and alkyl malonates to furnish the related products (Scheme 2). The structural diversity of reactants is summarized in Fig. 3, and the results are displayed in scheme 2 and Table 2. It is noteworthy that product (4) was produced when malononitrile was used, on the other hands, when ethyl cyanoacetate or methyl cyanoacetate was used, product (5) was produced. As Table 2 indicates, a variety of arylglyoxals, alkyl malonates, and 4-aminocoumarins were successfully applied in this process to afford the corresponding chromeno[4,3-b]pyrrol-3-yl derivatives. These reactions were completed within 1–3 h with good to excellent yields (87–97%). As shown in Table 2, substitutions of the methyl or methoxy moiety on arylglyoxal aromatic ring improved the reactivity compared to chlorine counterparts.

The synthesis of dihydrochromeno[4,3-b]pyrrol derivatives via the reaction between arylglyoxal (1), malono derivatives (2) and 4-amino coumarin derivatives (3) in ethanol under reflux conditions

Diversity elements employed for the synthesis of dihydrochromeno[4,3-b] pyrrole derivatives
aA mixture of 4-aminocoumarin derivative (1 mmol), arylglyoxal (1 mmol) and malono derivative (1 mmol) was stirred in EtOH (5 mL) at reflux to obtain the desired product 4 or 5.
b Isolated yields.
Based on the results, a plausible mechanism was proposed in (Scheme 3). Initially, under the Knoevenagel condensation reaction, arylglyoxal, and malono derivatives produced the intermediate 6. This intermediate then reacted with 4-amino coumarins to form intermediate 7. This intermediate was converted into 8 through the imine-enamine tautomerization, followed by N cyclization via attack to the C = O of arylglyoxal to produce 9. Then, hydrolysis of intermediate 9 gives product 4 when malononitrile was used as a starting material, and product 5 was formed when ethyl or methyl cyanoacetate were used as starting materials through decarboxylation of intermediate 10.

Proposed mechanism for the formation of products 4 and 5
In vitro α-glucosidase inhibitory activity
Various dihydrochromeno[4,3-b]pyrrol-3-yl derivatives (4a-5f) were synthesized and evaluated against α-glucosidase. The results are summarized in Table 3. The majority of the tested compounds displayed potent α-glucosidase inhibitory activity, with IC50 values in the range of 48.65 ± 0.01 to 733.83 ± 0.10 μM, when compared to acarbose (IC50 = 750.90 ± 0.14 μM) as the positive control. Amongst, compound 5e represented the most potent α-glucosidase inhibition with IC50 values of 48.65 ± 0.01 μM. To better understand the SAR, synthesized compounds were divided into two main categories, 4a-e (bearing CO-NH2 at R1) and 5a-f (containing H at the R1 position).

Evaluating the effect of R3 moiety on phenylpyrrole derivatives of 4a-e showed that unsubstituted derivative (4a) had an anti-a-glucosidase activity with around fivefold improvement in the inhibitory potency compared to the positive control. The presence of chlorine atom at 4- position of phenyl ring (4b) improved the potency compared to the unsubstituted one. Replacement of chlorine with methyl (4c) as a moderate electron-donating group resulted in a significant reduction in the inhibitory activity. It is interesting to point out that the presence of a strong electron-donating group such as meta-MeO (4d) on the aryl ring improved inhibition compared to 4c. The activity of 4e (R2 = Cl, R3 = 3-MeO) was slightly better than 4d (R2 = H, R3 = 3-MeO) indicated that the presence of chlorine group at R2 can improve the inhibitory activity.
Evaluation in the second category (compounds 5a-f) showed that unsubstituted phenyl ring with IC50 value of 282.97 µM (5a, R1 = H, R2 = H, R3 = H) had less activity in comparison with 4a (R1 = CO-NH2, R2 = H, R3 = H) counterpart. Disappointingly replacement of H at R2 in 5a with Cl (5b) moiety diminished the inhibitory activity completely. Unlike the previous group, the introduction of the methyl group at the 4-position of the phenyl ring (5c) improved inhibitory activity (IC50 = 131.03 ± 0.03 μM). The replacement of methyl (5c) with chlorine (5d) as halogen electron-withdrawing group significantly improved the α-glucosidase inhibition with around fivefold increased in potency compared to 5a. Compound 5e having 4-methoxy group showed the most potent inhibitory activity (IC50 = 48.65 μM) among all synthesized compounds. Unexpectedly changing the position of MeO from para to meta caused a significant decline in potency. This result indicated the position, as well as the type of substitution on the phenyl ring, was responsible for outstanding α-glucosidase inhibition.
Overall, it can be understood that the presence of para-chlorine or para-methoxy at R3 position on phenylpyrrole pendant had the most dominant role to improve activity against α-glucosidase.
Enzyme kinetic studies
According to Fig. 4a, the Lineweaver–Burk plot showed that the Km gradually increased and Vmax remained unchanged with increasing inhibitor concentration indicating a competitive inhibition. The results showed that 5e bond to the active site of the enzyme and competed with the substrate to bind to the active site. Furthermore, the plot of the Km versus different concentrations of 5e gave an estimate of the inhibition constant, Ki of 42.6 µM (Fig. 4b).

Kinetics of α-glucosidase inhibition by 5e. (a) The Lineweaver– Burk plot in the absence and presence of different concentrations of 5e; (b) the secondary plot between Km and various concentrations of 5e
Docking study
A molecular docking study was carried out to understand the possible interaction of 5e with the residues of the α-glucosidase active site. To validate the docking procedure, the reference drug, acarbose, was first docked into the binding site of α-glucosidase using AutoDock Tools version 1.5.6. The docking protocol was successful to regenerate the native co-crystallized orientation of the docked ligand with an RMSD value of 1.41 Å.
As can be seen in Fig. 5a, compound 5e fitted well in the active site of the enzyme. The interaction of the best-docked conformation of 5e with the residues of the active site is presented in Fig. 5b. Compound 5e constructed three hydrogen bonds with the active site. The nitrogen of the acetonitrile moiety formed a hydrogen bond with Tyr292 residue and the oxygen of chromenone ring also recorded two hydrogen bonds with Leu283 and Ala284 residues of the 5NN8 which confirmed the efficacy of 5e to induce the desired bioactivity. Also, chromenone ring formed pi–sigma and pi–alkyl interactions with Ala555. Pi-anion interaction between the pyrrole ring and Asp282 fixed compound 5e in the active site. At the other side of the molecule, methoxyphenyl moiety participated in two pi–anion interactions with Met519 and Asp616, two carbon–hydrogen bonds with Asp 616 plus pi–pi-t-shaped interaction with Trp481. Mentioned interactions might be beneficial to describe the promising efficacy of compound 5e against α-glucosidase.

The three-dimensional conformation of compound 5e docked into the active site (A). Three-dimensional orientation of compound 5e and important residues in the active site of 5NN8 (B). Hydrogen bonds are depicted in green dashed lines, Pi–Pi-T-shaped interactions are depicted in pink dashed lines, Pi–aryl interactions are depicted in dark pink dashed lines, Pi–anion interactions are depicted in orange dashed lines. Carbon–hydrogen bonds are depicted pale green dashed lines
Conclusions
In the present study, novel series of chromeno[4,3-b]pyrrol-3-yl derivatives were synthesized through ordinal Knoevenagel/Michael/intramolecular cyclization sequences in ethanol without using any catalyst. The present procedure has the advantage that not only the reaction is performed under neutral conditions but also the reactants can be mixed without any prior activation or modification. High atom economy, simple procedure in the excellent yields, easy workup procedure, and mild reaction conditions are the main advantages of this method.
All synthesized compounds were then evaluated as α-glucosidase inhibitors. Among them, compound 5e demonstrated the best inhibitory potency with an IC50 value of 48.65 μM which was 19 times more potent than acarbose as a standard inhibitor. Moreover, limited SARs of chromeno[4,3-b]pyrrol-3-yl derivatives showed that the presence of para-methoxy or para-chlorine substituent on phenyl ring improved the inhibitory activity. Enzyme kinetic studies showed that compound 5e is a competitive inhibitor with a Ki of 42.6 µM. Furthermore, the docking study was demonstrated that compound 5e binds to the active site of the α-glucosidase through both hydrophobic and hydrogen bond interactions. The current study provides a new class of compounds for the development of novel α-glucosidase inhibitors.
Experimental section
Chemistry
All chemicals were purchased from Merck or Fluka chemical companies. The 1H NMR (500 MHZ) and 13C NMR (125 MHZ) were run on a Varian—Inova 500. Melting points were recorded on a Stuart Scientific Apparatus SMP3 (UK) in open capillary tubes. Elemental analyses for C, H, and N were performed using a Thermo Finnigan FLASH EA. Reaction progress was screened by TLC using silicagel polygram SIL G/UV254 plates. Mass spectra were recorded on an Agilent Technology (HP) 5973 mass spectrometer operating at an ionization potential of 70 eV.
General procedure for the synthesis of arylglyoxal
18.5 g of selenium dioxide was dissolved in 100 mL of dioxane by warming to 50° C and stirring until all was in solution. To this solution was added 23 g of acetophenone derivatives and the reaction was refluxed for 4 h. The hot solution was decanted from the solid selenium, and the dioxane and water were removed by distillation through a short column at atmospheric pressure.
General procedure for the synthesis of chromeno[4,3-b]pyrrol-3-yl derivatives 4a-e, 5a-f
A mixture of arylglyoxal 1 (1 mmol), malono derivatives 2 (1 mmol), and 4-aminocoumarin derivatives 3 (1 mmol) was stirred in EtOH reflux (5 mL), and the solution was heated at reflux for the time given in Table 2. Upon completion as monitored by TLC, the reaction mixture was cooled, the precipitate formed was filtered off and washed on the filter funnel with a small amount of ethanol to give pure products.
2-cyano-2-(4-oxo-2-phenyl-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)acetamide (4a):
White powder, m.p > 300 °C. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 5.47 (s, 1H), 7.38 (s, 1H), 7.43 (dd, J = 1.5 Hz, 7.5 Hz, 1H), 7.49–7.56 (m, 3H), 7.57–7.66 (m, 5H), 8.22 (dd, J = 1.5 Hz, 7.5 Hz, 1H), 12.97 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 35.55, 107.64, 109.29, 113.83, 117.42, 122.22, 124.91, 129.27, 129.33, 129.41, 129.56, 129.78, 130.24, 136.12, 136.81, 151.82, 158.50, 165.90. EI-MS (70 eV): m/z (%) = 340 (M+, 9), 325 (47), 300 (100). Anal. calcd for C20H13N3O3: C, 69.97; H, 3.82; N, 12.24%. Found: C, 69.97; H, 3.82; N, 12.24%.
2-(2-(4-chlorophenyl)-4-oxo-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)-2-cyanoacetamide (4b):
White powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 6.43 (s, 1H), 7.41–7.45 (m, 1H), 7.49- 7.67 (m, 5H), 7.71–7.74 (m, 2H), 8.16 (dd, J = 1.5 Hz, 7.5 Hz, 1H), 13.31 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 20.03, 104.86, 106.92, 113.23, 117.53, 122.35, 125.05, 127.91, 129.71, 130.32, 131.12, 131.37, 134.89, 136.00, 136.90, 152.01, 157.77. EI-MS (70 eV): m/z (%) = 379 (M+ + 2, 0.2), 377 (M+, 0.6), 359 (100). Anal. calcd for C20H12ClN3O3: C, 63.59; H, 3.20; N, 11.12%. Found: C, 63.58; H, 3.20; N, 11.10%.
2-cyano-2-(4-oxo-2-(p-tolyl)-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)acetamide (4c):
White powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.41 (s, 3H), 5.46 (s, 1H), 7.37–7.43 (m, 4H), 7.47–7.59 (m, 5H), 8.21 (d, J = 7.5 Hz, 1H), 12.90 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 21.39, 35.59, 107.63, 108.94, 113.37, 113.84, 117.37, 117.42, 122.18, 124.85, 127.40, 129.16, 129.66, 129.83, 130.25, 135.97, 136.89, 138.95, 151.77, 158.54, 165.97. EI-MS (70 eV): m/z (%) = 357 (M+, 1), 339 (7), 314 (100). Anal. calcd for C21H15N3O3: C, 70.58; H, 4.23; N, 11.76%. Found: 70.55; H, 4.20; N, 11.75%.
2-cyano-2-(2-(3-methoxyphenyl)-4-oxo-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)acetamide (4d):
White powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 3.84 (s, 3H), 5.52 (s, 1H), 7.07 (d, J = 7.5 Hz, 1H), 7.19–7.22 (m, 2H), 7.37–7.43 (m, 2H), 7.47–7.53 (m, 3H), 7.59 (s, 1H), 8.21 (d, J = 7.5 Hz, 1H), 12.94 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 36.53, 56.63, 108.65, 110.34, 114.76, 115.70, 116.10, 118.39, 120.80, 122.36, 123.23, 125.89, 130.79, 131.44, 132.42, 137.04, 137.59, 152.77, 159.55, 160.72, 166.95. EI-MS (70 eV): m/z (%) = 373 (M+, 15), 356 (59), 330 (100). Anal. calcd for C21H15N3O4: C, 67.56; H, 4.05; N, 11.25%. Found: C, 67.51; H, 4.10; N, 11.25%.
2-(8-chloro-2-(3-methoxyphenyl)-4-oxo-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)-2-cyanoacetamide (4e):
White powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 3.83 (s, 3H), 5.51 (s, 1H), 7.03–7.20 (m, 3H), 7.39–7.50 (m, 4H), 7.62 (s, 1H), 8.19 (d, J = 7.5 Hz, 1H), 12.96 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 35.54, 55.66, 107.66, 109.37, 113.78, 114.74, 115.12, 117.35, 117.40, 121.39, 122.24, 124.89, 129.78, 130.43, 131.43, 136.04, 136.59, 151.79, 158.55, 159.73, 165.93. EI-MS (70 eV): m/z (%) = 409 (M+ + 2, 0.2), 407 (M+, 0.6), 373 (15), 330 (100). Anal. calcd for C21H14ClN3O4: C, 61.85; H, 3.46; N, 10.30%. Found: C, 61.82; H, 3.43; N, 10.30%.
2-(4-oxo-2-phenyl-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)acetonitrile (5a):
White powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 4.15 (s, 2H), 7.40–7.44 (m, 1H), 7.46–7.54 (m, 3H), 7.58–7.68 (m, 4H), 8.21 (dd, J = 1.5 Hz, 7.5 Hz, 1H), 12.91 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 13.89, 107.69, 108.67, 113.78, 117.43, 118.93, 122.24, 124.84, 128.56, 129.10, 129.57, 129.70, 130.42, 135.41, 135.89, 151.82, 158.57. EI-MS (70 eV): m/z (%) = 300 (M+, 100), 271 (8), 255 (7). Anal. calcd for C19H12N2O2: C, 75.99; H, 4.03; N, 9.33%. Found: C, 76; H, 4.02; N, 9.30%.
2-(8-chloro-4-oxo-2-phenyl-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)acetonitrile (5b):
White powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 4.15 (s, 2H), 7.40–7.44 (m, 1H), 7.48–7.54 (m, 3H), 7.61–7.68 (m, 3H), 8.21 (dd, J = 1.5 Hz, 7.5 Hz, 1H), 12.91 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 13.89, 107.68, 108.66, 113.76, 117.41, 118.93, 122.23, 124.82, 128.55, 129.09, 129.56, 129.68, 130.42, 135.40, 135.88, 151.81, 158.56. EI-MS (70 eV): m/z (%) = 336 (M+ + 2, 0.2), 334 (M+, 0.7), 300 (M+, 100). Anal. calcd for C19H11ClN2O2: C, 68.17; H, 3.31; N, 8.37%. Found: C, 68.17; H, 3.31; N, 8.37%.
2-(4-oxo-2-(p-tolyl)-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)acetonitrile (5c):
White powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 2.42 (s, 3H), 4.13 (s, 2H), 7.40–7.45 (m, 3H), 7.46–7.53 (m, 2H), 7.55–7.57(m, 2H), 8.21 (dd, J = 1.5 Hz, 7.5 Hz, 1H), 12.84 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 13.89, 21.34, 107.65, 108.22, 113.78, 117.36, 118.96, 122.19, 124.76, 127.57, 128.37, 129.55, 130.09, 135.47, 135.69, 138.64, 151.76, 158.57. EI-MS (70 eV): m/z (%) = 314 (M+, 100). Anal. calcd for C20H14N2O2: C, 76.42; H, 4.49; N, 8.91%. Found: C, 76.40; H, 4.45; N, 8.90%.
2-(2-(4-chlorophenyl)-4-oxo-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)acetonitrile (5d):
Cream powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 4.17 (s, 2H), 7.40–7.43 (m, 1H), 7.45–7.54 (m, 2H), 7.65–7.74 (m, 4H), 8.185 (d, J = 7.5 Hz, 1H), 12.93 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 13.78, 107.73, 109.21, 113.67, 117.44, 118.79, 122.26, 124.86, 129.27, 129.59, 129.59, 129.81, 130.25, 130.26, 133.78, 134.07, 136.06, 151.84, 158.50. EI-MS (70 eV): m/z (%) = 336 (M+ + 2, 35), 334 (M+, 100). Anal. calcd for C19H11ClN2O2: C, 68.17; H, 3.31; N, 8.37%. Found: C, 68.14; H, 3.28; N, 8.4%.
2-(2-(4-methoxyphenyl)-4-oxo-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)acetonitrile (5e):
Cream powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 3.84 (s, 3H), 4.09 (s, 2H), 7.16 (d, J = 7.5 Hz, 2H), 7.37- 7.41 (m, 1H), 7.44–7.51 (m, 2H), 7.57–7.59 (m, 2H), 8.17 (d, J = 7.5 Hz, 1H), 12.79 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 13.86, 55.80, 107.57, 107.73, 113.81, 115.01, 115.01, 117.37, 119.02, 122.13, 122.74, 124.77, 129.48, 129.95, 129.95, 135.46, 135.50, 151.73, 158.59, 159.97. EI-MS (70 eV): m/z (%) = 330 (M+, 100), 315 (26), 287 (28). Anal. calcd for C20H14N2O3: C, 72.72; H, 4.27; N, 8.48%. Found: C, 72.7; H, 4.25; N, 8.45%.
2-(2-(3-methoxyphenyl)-4-oxo-1,4-dihydrochromeno[4,3-b]pyrrol-3-yl)acetonitrile (5f):
White powder, m.p > 300 °C, 1H NMR (DMSO-d6, 500 MHz) δ (ppm): 3.86 (s, 3H), 4.14 (s, 2H), 7.06–7.09 (m, 1H), 7.19 (s, 1H), 7.22 (d, J = 7.5 Hz, 1H), 7.39–7.54 (m, 4H), 8.20 (d, J = 7.5 Hz, 1H), 12.87 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ (ppm): 13.93, 55.75, 107.68, 108.79, 113.74, 114.10, 114.63, 117.41, 118.97, 120.73, 122.27, 124.81, 129.70, 130.76, 131.64, 135.24, 135.83, 151.82, 158.56, 160.04. EI-MS (70 eV): m/z (%) = 330 (M+, 100), 287 (30). Anal. calcd for C20H14N2O3: C, 72.72; H, 4.27; N, 8.48%. Found: C, 72.7; H, 4.25; N, 8.45%.
α-glucosidase inhibition assay
α-glucosidase inhibition assay was performed exactly according to the previously reported procedure [20, 51,52,53].
Enzyme kinetic studies
The mode of inhibition of the most active compound (5e), identified with the lowest IC50, was investigated against α-glucosidase at different concentrations of p-nitrophenyl α-D-glucopyranoside (2–10 mM) as substrate in the absence and presence of 5e at different concentrations (0, 12, 25, and 50 µM). A Lineweaver–Burk plot was generated to identify the type of inhibition, and the Michaelis–Menten constant (Km) value was determined from the plot between reciprocal of the substrate concentration (1/[S]) and reciprocal of enzyme rate (1/V) over various inhibitor concentrations. The experimental inhibitor constant (Ki) value was constructed by secondary plots of the inhibitor concentration [I] versus Km [52].
Molecular docking
The 3D structure of α-glucosidase with PDB ID: 5NN8 (EC: 3.2.1.20, resolution: 2.45 Å) was downloaded from the Brookhaven protein database (https://www.rcsb.org/structure/5NN8). Docking studies were performed using AutoDock Tools (version1.5.6). The 3D structure of the selected compound was generated and energy minimized using hyperchem software and then converted to pdbqt coordinate via Autodock Tools. Before docking, the water molecules and the inhibitor were removed from the protein. Then, using AutoDock Tools, polar hydrogen atoms were added, and Kollman charges were assigned. The active site is defined by the ligand of the protein crystal structure. The dimensions of the active site box were set at 60 × 60 × 60 Å with flexible ligand dockings approach. The docked system was carried out by 50 runs of the AUTODOCK search by the Lamarckian genetic algorithm (LGA). The best position of 5e was selected for analyzing the interactions against α-glucosidase. The results were visualized using Discovery Studio 2016 Client [22].
References
Hasaninejad A, Zare A, Shekouhy M (2011) Highly efficient synthesis of triazolo [1, 2-a] indazole-triones and novel spiro triazolo [1, 2-a] indazole-tetraones under solvent-free conditions. Tetrahedron 67(2):390–400. https://doi.org/10.1016/j.tet.2010.11.029
Zolfigol MA, Khazaei A, Zare A, Mokhlesi M, Hekmat-Zadeh T, Hasaninejad A, Derakhshan-Panah F, Moosavi-Zare AR, Keypour H, Dehghani-Firouzabadi AA (2012) Solid-supported sulfonic acid-containing catalysts efficiently promoted one-pot multi-component synthesis of β-acetamido carbonyl compounds. J Chem Sci 124(2):501–508. https://doi.org/10.1007/s12039-011-0210-4
Zare A, Khanivar R, Hatami M, Mokhlesi M, Zolfigol MA, Moosavi-Zare AR, Hasaninejad A, Khazaei A, Khakyzadeh V (2012) Efficient Synthesis of 12-Aryl-8, 9, 10, 12-tetrahydrobenzo [α]-xanthen-11-ones using Ionic Liquid Pyrazinium Di (hydrogen sulfate){Py (HSO4) 2} as a Novel, Green and Homogeneous Catalyst. J Mex Chem Soc 56(4):389–397
Edraki N, Firuzi O, Fatahi Y, Mahdavi M, Asadi M, Emami S, Divsalar K, Miri R, Iraji A, Khoshneviszadeh M, Firoozpour L, Shafiee A, Foroumadi A (2015) N-(2-(Piperazin-1-yl)phenyl)arylamide Derivatives as β-Secretase (BACE1) Inhibitors: Simple Synthesis by Ugi Four-Component Reaction and Biological Evaluation. Arch Pharm 348(5):330–337. https://doi.org/10.1002/ardp.201400322
Hasaninejad A, Firoozi S, Mandegani F (2013) An efficient synthesis of novel spiro[benzo[c]pyrano[3,2-a]phenazines] via domino multi-component reactions using l-proline as a bifunctional organocatalyst. Tetrahedron Lett 54(22):2791–2794. https://doi.org/10.1016/j.tetlet.2013.03.073
Wondafrash DZ, Desalegn TZ, Yimer EM, Tsige AG, Adamu BA, Zewdie KA (2020) Potential effect of hydroxychloroquine in diabetes mellitus: a systematic review on preclinical and clinical trial studies. J Diabetes Res 5(6):1–10. https://doi.org/10.1155/2020/5214751
Khan MAB, Hashim MJ, King JK, Govender RD, Mustafa H, Al Kaabi J (2020) Epidemiology of Type 2 Diabetes - Global Burden of Disease and Forecasted Trends. J Epidemiol Glob Health 10(1):107–111. https://doi.org/10.2991/jegh.k.191028.001
E. Blackburn, The Emerging Role of Pancreatic β-Cell Primary Cilia in Diabetes Mellitus, 4(28) (2021). https://opencommons.uconn.edu/srhonors_theses/777.
Alssema M, Ruijgrok C, Blaak EE, Egli L, Dussort P, Vinoy S, Dekker JM, Denise Robertson M (2021) Effects of alpha-glucosidase-inhibiting drugs on acute postprandial glucose and insulin responses: a systematic review and meta-analysis. Nutrition Diabetes. https://doi.org/10.1038/s41387-021-00152-5
Tundis R, Loizzo M, Menichini F (2010) Natural products as α-amylase and α-glucosidase inhibitors and their hypoglycaemic potential in the treatment of diabetes: an update. Mini Rev Med Chem 10(4):315–331. https://doi.org/10.2174/138955710791331007
Khursheed R, Singh SK, Wadhwa S, Kapoor B, Gulati M, Kumar R, Ramanunny AK, Awasthi A, Dua K (2019) Treatment strategies against diabetes: Success so far and challenges ahead. Eur J Pharmacol 862:172625. https://doi.org/10.1016/j.ejphar.2019.172625
Hakamata W, Kurihara M, Okuda H, Nishio T, Oku T (2009) Design and screening strategies for α-glucosidase inhibitors based on enzymological information. Curr Top Med Chem 9(1):3–12. https://doi.org/10.2174/156802609787354306
Menteşe E, Baltaş N, Bekircan O (2019) Synthesis and kinetics studies of N′-(2-(3,5-disubstituted-4H-1,2,4-triazol-4-yl)acetyl)-6/7/8-substituted-2-oxo-2H-chromen-3-carbohydrazide derivatives as potent antidiabetic agents. Arch Pharm 352(12):1900227. https://doi.org/10.1002/ardp.201900227
Bischoff H (1995) The mechanism of alpha-glucosidase inhibition in the management of diabetes. Clin Invest Med 18(4):303–311
Ríos JL, Francini F, Schinella GR (2015) natural products for the treatment of type 2 diabetes mellitus. Planta Med 81(12–13):975–994. https://doi.org/10.1055/s-0035-1546131
Wang L, Tan N, Wang H, Hu J, Diwu W, Wang X (2020) A systematic analysis of natural α-glucosidase inhibitors from flavonoids of Radix scutellariae using ultrafiltration UPLC-TripleTOF-MS/MS and network pharmacology. BMC Complement Med Ther. https://doi.org/10.1186/s12906-020-2871-3
J. Djelmi, G. Desoye, Diabetology of pregnancy, Karger Medical and Scientific Publishers 2005.
V. Mohan, R. Unnikrishnan, World Clinics: Diabetology-Type 2 Diabetes Mellitus, JP Medical Ltd2014.
Chaudhry F, Shahid W, Al-Rashida M, Ashraf M, Munawar MA, Khan MA (2021) Synthesis of imidazole-pyrazole conjugates bearing aryl spacer and exploring their enzyme inhibition potentials. Bioorg Chem 108:104686. https://doi.org/10.1016/j.bioorg.2021.104686
Azimi F, Ghasemi JB, Azizian H, Najafi M, Faramarzi MA, Saghaei L, Sadeghi-Aliabadi H, Larijani B, Hassanzadeh F, Mahdavi M (2021) Design and synthesis of novel pyrazole-phenyl semicarbazone derivatives as potential α-glucosidase inhibitor: Kinetics and molecular dynamics simulation study. Int J Biol Macromol 166:1082–1095. https://doi.org/10.1016/j.ijbiomac.2020.10.263
Sherafati M, Mirzazadeh R, Barzegari E, Mohammadi-Khanaposhtani M, Azizian H, Sadegh Asgari M, Hosseini S, Zabihi E, Mojtabavi S, Ali Faramarzi M, Mahdavi M, Larijani B, Rastegar H, Hamedifar H, Hamed Hajimiri M (2021) Quinazolinone-dihydropyrano[3,2-b]pyran hybrids as new α-glucosidase inhibitors: Design, synthesis, enzymatic inhibition, docking study and prediction of pharmacokinetic. Bioorganic Chemistry. https://doi.org/10.1016/j.bioorg.2021.104703
Shareghi-Boroujeni D, Iraji A, Mojtabavi S, Faramarzi MA, Akbarzadeh T, Saeedi M (2021) Synthesis, in vitro evaluation, and molecular docking studies of novel hydrazineylideneindolinone linked to phenoxymethyl-1,2,3-triazole derivatives as potential α-glucosidase inhibitors. Bioorg Chem 111:104869. https://doi.org/10.1016/j.bioorg.2021.104869
Santos CMM, Freitas M, Fernandes E (2018) A comprehensive review on xanthone derivatives as α-glucosidase inhibitors. Eur J Med Chem 157:1460–1479. https://doi.org/10.1016/j.ejmech.2018.07.073
Sari S, Barut B, Özel A, Saraç S (2020) Discovery of potent α-glucosidase inhibitors through structure-based virtual screening of an in-house azole collection. Chem Biol Drug Des. https://doi.org/10.1111/cbdd.13805
Suzuki M, Nakagawa-Goto K, Nakamura S, Tokuda H, Morris-Natschke SL, Kozuka M, Nishino H, Lee K-H (2006) Cancer preventive agents Part 5 anti-tumor-promoting effects of coumarins and related compounds on Epstein-Barr virus activation and two-stage mouse skin carcinogenesis. Pharmaceutical biology. https://doi.org/10.1080/13880200600686491
Kayser O, Kolodziej H (1999) Antibacterial activity of simple coumarins: structural requirements for biological activity. Zeitschrift für Naturforschung C 54(3–4):169–174. https://doi.org/10.1515/znc-1999-3-405
Garazd YL, Kornienko E, Maloshtan L, Garazd M, Khilya V (2005) Modified coumarins. 17. Synthesis and anticoagulant activity of 3, 4-cycloannelated coumarin D-glycopyranosides. Chem nat comp. https://doi.org/10.1007/s10600-005-0194-8
Kontogiorgis CA, Hadjipavlou-Litina DJ (2005) Synthesis and antiinflammatory activity of coumarin derivatives. J Med Chem 48(20):6400–6408. https://doi.org/10.1021/jm0580149
Almerico AM, Diana P, Barraja P, Dattolo G, Mingoia F, Loi AG, Scintu F, Milia C, Puddu I, La Colla P (1998) Glycosidopyrroles Part 1 Acyclic derivatives: 1-(2-hydroxyethoxy) methylpyrroles as potential anti-viral agents. Il Farmaco. https://doi.org/10.1016/s0014-827x(97)00002-5
Soenen DR, Hwang I, Hedrick MP, Boger DL (2003) Multidrug resistance reversal activity of key ningalin analogues. Bioorg Med Chem Lett 13(10):1777–1781. https://doi.org/10.1016/s0960-894x(03)00294-4
Dubuffet T, Newman-Tancredi A, Cussac D, Audinot V, Loutz A, Millan MJ, Lavielle G (1999) Novel benzopyrano [3, 4-c] pyrrole derivatives as potent and selective dopamine D3 receptor antagonists. Bioorg Med Chem Lett 9(14):2059–2064. https://doi.org/10.1016/s0960-894x(99)00312-1
Ridley CP, Reddy MVR, Rocha G, Bushman FD, Faulkner DJ (2002) Total synthesis and evaluation of lamellarin α 20-Sulfate analogues. Bioorg Med Chem 10(10):3285–3290. https://doi.org/10.1016/S0968-0896(02)00237-7
Dubuffet T, Muller O, Simonet SS, Descombes J-J, Laubie M, Verbeuren TJ, Lavielle G (1996) Synthesis of new 3,4-disubstituted pyrrolidines as thromboxane A2/prostaglandin H2 (TP) receptor antagonists. Bioorg Med Chem Lett 6(4):349–352. https://doi.org/10.1016/0960-894X(96)00021-2.SayanMukherjee,ChemistrySelect2018,3,1537-1544
Mukherjee S, Sarkar S, Pramanik A (2018) Synthesis of functionalized pyrrole fused coumarins under solvent-free conditions using magnetic nanocatalyst and a new route to polyaromatic indolocoumarins. ChemistrySelect 3:1537–1544. https://doi.org/10.1002/slct.201703146
Yang X, Li Ch, Zhang F, Qi Ch (2021) An efficient domino strategy for synthesis of 3-substituted 4-oxo-4,5-dihydro-1H-pyrrolo[3,2-c]pyridine derivatives in water. Mol Divers. https://doi.org/10.1007/s11030-021-10294-4
Mohammadi Ziarani Gh, Moradi R, Ahmadi T, Gholamzadeh P (2019). Mol Divers. https://doi.org/10.1007/s11030-019-09918-7
Dai Ch, Xie Z, Qing X, Luo N, Wang C (2019) DBU-mediated annulation of 2-aryl-3-nitro-2H-chromenes with 1,3-cyclohexanediones for the synthesis of benzofuro[2,3-c] chromenone derivatives. Mol Divers. https://doi.org/10.1007/s11030-019-09939-2
Yahyavi H, Heravi MM, Mahdavi M, Foroumadi A (2018) Iodine-catalyzed tandem oxidative coupling reaction: A one-pot strategy for the synthesis of new coumarin-fused pyrroles. Tetrahedron Lett 59(2):94–98. https://doi.org/10.1016/j.tetlet.2017.11.055
Saha M, Pradhan K, Das AR (2016) Facile and eco-friendly synthesis of chromeno[4,3-b]pyrrol-4(1H)-one derivatives applying magnetically recoverable nano crystalline CuFe2O4 involving a domino three-component reaction in aqueous media. RSC Adv 6(60):55033–55038. https://doi.org/10.1039/c6ra06979g
Hasaninejad A, Mojikhalifeh S, Beyrati M (2018) Highly efficient, catalyst-free, one-pot, pseudo five-component synthesis of novel pyrazoline-containing Schiff bases, metal complexes formation and computational studies via DFT method. Appl Organomet Chem 32(7):e4380. https://doi.org/10.1002/aoc.4380
Mojikhalifeh S, Hasaninejad A (2018) Highly efficient, catalyst-free, one-pot, pseudo-seven-component synthesis of novel poly-substituted pyrazolyl-1,2-diazepine derivatives. Organic Chemistry Frontiers 5(9):1516–1521. https://doi.org/10.1039/C8QO00210J
Beyrati M, Forutan M, Hasaninejad A, Rakovský E, Babaei S, Maryamabadi A, Mohebbi G (2017) One-pot, four-component synthesis of spiroindoloquinazoline derivatives as phospholipase inhibitors. Tetrahedron 73(34):5144–5152. https://doi.org/10.1016/j.tet.2017.07.005
Maryamabadi A, Hasaninejad A, Nowrouzi N, Mohebbi G (2017) Green synthesis of novel spiro-indenoquinoxaline derivatives and their cholinesterases inhibition activity. Bioorg Med Chem 25(7):2057–2064. https://doi.org/10.1016/j.bmc.2017.02.017
Chaudhry F, Naureen S, Huma R, Shaukat A, Al-Rashida M, Asif N, Ashraf M, Munawar MA, Khan MA (2017) In search of new α-glucosidase inhibitors: Imidazolylpyrazole derivatives. Bioorg Chem 71:102–109. https://doi.org/10.1016/j.bioorg.2017.01.017
Wang G, He D, Li X, Li J, Peng Z (2016) Design, synthesis and biological evaluation of novel coumarin thiazole derivatives as α-glucosidase inhibitors. Bioorg Chem 65:167–174. https://doi.org/10.1016/j.bioorg.2016.03.001
Salar U, Taha M, Khan KM, Ismail NH, Imran S, Perveen S, Gul S, Wadood A (2016) Syntheses of new 3-thiazolyl coumarin derivatives, in vitro α-glucosidase inhibitory activity, and molecular modeling studies. Eur J Med Chem 122:196–204. https://doi.org/10.1016/j.ejmech.2016.06.037
Witte JF, McClard RW (1991) Synthesis of a potent α-glucosidase inhibitor epimeric to fr 900483. Tetrahedron Lett 32(32):3927–3930. https://doi.org/10.1016/0040-4039%2891%2980591-S
Jadhav NC, Pahelkar AR, Desai NV, Telvekar VN (2017) Design, synthesis and molecular docking study of novel pyrrole-based α-amylase and α-glucosidase inhibitors. Med Chem Res 26(10):2675–2691. https://doi.org/10.1007/s00040-017-1965-z
Chen S, Yong T, Xiao C, Su J, Zhang Y, Jiao C, Xie Y (2018) Pyrrole alkaloids and ergosterols from Grifola frondosa exert anti-α-glucosidase and anti-proliferative activities. Journal of functional foods 43:196–205. https://doi.org/10.1016/j.jff.2018.02.007
Tiffany BD, Wright JB, Moffett RB, Heinzelman RV, Strube RE, Aspergren BD, Lincoln EH, White JL (1957) Antiviral compounds I Aliphatic glyoxals α-hydroxyaldehydes and related compounds. J Am Chem Soc. https://doi.org/10.1021/JA01564A042
Mohammadi-Khanaposhtani M, Rezaei S, Khalifeh R, Imanparast S, Faramarzi MA, Bahadorikhalili S, Safavi M, Bandarian F, Esfahani EN, Mahdavi M (2018) Design, synthesis, docking study, α-glucosidase inhibition, and cytotoxic activities of acridine linked to thioacetamides as novel agents in treatment of type 2 diabetes. Bioorg Chem 80:288–295. https://doi.org/10.1016/j.bioorg.2018.06.035
Nikookar H, Mohammadi-Khanaposhtani M, Imanparast S, Faramarzi MA, Ranjbar PR, Mahdavi M, Larijani B (2018) Design, synthesis and in vitro α-glucosidase inhibition of novel dihydropyrano [3, 2-c] quinoline derivatives as potential anti-diabetic agents. Bioorg Chem 77:280–286. https://doi.org/10.1016/j.bioorg.2018.01.025
Azimi F, Azizian H, Najafi M, Hassanzadeh F, Sadeghi-Aliabadi H, Ghasemi JB, Faramarzi MA, Mojtabavi S, Larijani B, Saghaei L (2021) Design and synthesis of novel quinazolinone-pyrazole derivatives as potential α-glucosidase inhibitors: Structure-activity relationship molecular modeling and kinetic study. Bioorganic Chem. https://doi.org/10.1016/j.bioorg.2021.105127
Acknowledgements
The authors thank Persian Gulf University Research Council for the financial support of this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Karami, M., Hasaninejad, A., Mahdavi, H. et al. One-pot multi-component synthesis of novel chromeno[4,3-b]pyrrol-3-yl derivatives as alpha-glucosidase inhibitors. Mol Divers 26, 2393–2405 (2022). https://doi.org/10.1007/s11030-021-10337-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10337-w