
Abstract
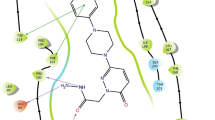
Parkinson’s disease (PD) is a degenerative disorder of the CNS, characterized by cerebral depletion of dopamine (DA), hence one of the approaches to delay the depletion of DA is to inhibit its oxidative deamination. Monoamine oxidases (MAO) carry out the oxidative deamination of monoamines like DA. These are intracellular enzymes, located on the outer mitochondrial membrane. MAO-A and MAO-B are the two subtypes of which MAO-B is the most predominant enzyme and is commonly found in the brain. Inhibition of the MAO-B enzyme boosts the effect of both endogenous and exogenous DA. In this study, we have carried out crystal structure analysis and structure-based design of MAO-B inhibitors. We also performed molecular dynamics, flexible docking, induced-fit docking and ADME prediction of the newly designed compounds.









Similar content being viewed by others

References
Savitt JM, Dawson VL, Dawson TM (2006) Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Investig 116:1744. doi:10.1172/JCI29178
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39:889–909. doi:10.1016/S0896-6273(03)00568-3
de Lau LML, Breteler M (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5:525–535. doi:10.1016/S1474-4422(06)70471-9
Bortolato M, Chen K, Shih JC (2008) Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev 60:1527–1533. doi:10.1016/j.addr.2008.06.002
Arias-Carrion O, P ppel E (2007) Dopamine, learning, and reward-seeking behavior. Acta Neurobiol Exp 67:481–488
Factor SA (2008) Current status of symptomatic medical therapy in Parkinson’s disease. Neurotherapeutics 5:164–180. doi:10.1016/j.nurt.2007.12.001
Braun GH, Jorge DM, Ramos HP, Alves RM, Da Silva VB, Giuliatti S, Sampaio SV, Taft CA, Silva CH (2008) Molecular dynamics, flexible docking, virtual screening, ADMET predictions, and molecular interaction field studies to design novel potential MAO-B inhibitors. J Biomol Struct Dyn 25:347–355
De Lano W (2002) PyMOL version 0.99. DeLano Scientific, San Carlos, CA
Sybyl T 7.0. Tripos Inc. St Louis, MO
Bohm HJ (1992) The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput Aided Mol Des 6:61–78. doi:10.1007/BF00124387
Studio D (2009) Version 2.5. Accelrys Inc, San Diego
Wang R, Gao Y, Lai L (2000) LigBuilder: a multi-purpose program for structure-based drug design. J Mol Model 6:498–516. doi:10.1007/s0089400060498
Lindahl E, Hess B, Van Der Spoel D (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model 7:306–317. doi:10.1007/s008940100045
Berendsen HJC, van der Spoel D, van Drunen R (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Comput Phys Commun 91:43–56. doi:10.1016/0010-4655(95)00042-E
Schuttelkopf AW, Van Aalten DMF (2004) PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D 60:1355–1363. doi:10.1107/S0907444904011679
Berendsen HJC, Postma JPM, Van Gunsteren WF, Hermans J (1981) Interaction models for water in relation to protein hydration. Intermol Forces 331:331–342
Miyamoto S, Kollman PA (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem 13:952–962. doi:10.1002/jcc.540130805
Turner PJ (2005) XMGRACE, version 5.1. 19. Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38. doi:10.1016/0263-7855(96)00018-5
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Daniel T, Repasky MP, Knoll EH, Shelley M, Perry JK (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47:1739–1749. doi:10.1021/jm0306430
Schrödinger (2009) Glide, v. 5.5. Schrödinger, New York
Morris G (2002) SYBYL software, version 7.1. Tripos Associates, St. Louis
Schrödinger Suite (2009) Protein Preparation Wizard; Epik version 2.0 S, LLC., New, York, N., 2009; Impact version 5.5, Schrödinger, LLC., New York, NY, 2009; Prime version, 2.1, S., LLC., New York, NY, 2009
LigPrep (2009) Schrödinger, New York
Schrödinger Suite (2009) Induced fit docking protocol; glide version 5.5. Schrödinger, New York
QikProp v. 3.2 Schrödinger, New York (2009)
Binda C, Li M, Hubálek F, Restelli N, Edmondson DE, Mattevi A (2003) Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc Natl Acad Sci USA 100:9750. doi:10.1073/pnas.1633804100
DeLano WL (2002) The PyMOL user’s manual. DeLano Scientific, San Carlos, CA
MOE 2007.09 Schrödinger, MO (2009)
Acknowledgments
The authors would like to thank the Department of Science and Technology (DST) and the Council of Scientific and Industrial Research (CSIR), New Delhi, India for their financial assistance.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Kare, P., Bhat, J. & Sobhia, M.E. Structure-based design and analysis of MAO-B inhibitors for Parkinson’s disease: using in silico approaches. Mol Divers 17, 111–122 (2013). https://doi.org/10.1007/s11030-012-9420-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-012-9420-z