
Abstract
Short-chain enoyl-CoA hydratase deficiency (ECHS1D) is a rare congenital metabolic disorder that follows an autosomal recessive inheritance pattern. It is caused by mutations in the ECHS1 gene, which encodes a mitochondrial enzyme involved in the second step of mitochondrial β-oxidation of fatty acids. The main characteristics of the disease are severe developmental delay, regression, seizures, neurodegeneration, high blood lactate, and a brain MRI pattern consistent with Leigh syndrome. Here, we report three patients belonging to a consanguineous family who presented with mitochondrial encephalomyopathy. Whole-exome sequencing revealed a new homozygous mutation c.619G > A (p.Gly207Ser) at the last nucleotide position in exon 5 of the ECHS1 gene. Experimental analysis showed that normal ECHS1 pre-mRNA splicing occurred in all patients compared to controls. Furthermore, three-dimensional models of wild-type and mutant echs1 proteins revealed changes in catalytic site interactions, conformational changes, and intramolecular interactions, potentially disrupting echs1 protein trimerization and affecting its function. Additionally, the quantification of mtDNA copy number variation in blood leukocytes showed severe mtDNA depletion in all probands.







Similar content being viewed by others

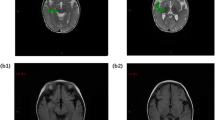

Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Abdenur JE, Sowa M, Simon M et al (2020) Medical nutrition therapy in patients with HIBCH and ECHS1 defects: clinical and biochemical response to low valine diet. Mol Genet Metab Rep 24:100617. https://doi.org/10.1016/j.ymgmr.2020.100617
Al Mutairi F, Shamseldin HE, Alfadhel M et al (2017) A lethal neonatal phenotype of mitochondrial short-chain enoyl-CoA hydratase-1 deficiency. Clin Genet 91:629–633. https://doi.org/10.1111/cge.12891
Aoto Y, Horinouchi T, Yamamura T et al (2022) Last nucleotide substitutions of COL4A5 exons cause aberrant splicing. Kidney Int Rep 7:108–116. https://doi.org/10.1016/j.ekir.2021.10.012
Bell AF, Wu J, Feng Y, Tonge PJ (2001) Involvement of glycine 141 in substrate activation by enoyl-CoA hydratase. Biochemistry 40:1725–1733. https://doi.org/10.1021/bi001733z
Bromberg Y, Rost B (2007) SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res 35:3823–3835. https://doi.org/10.1093/nar/gkm238
Burgin HJ, Crameri JJ, Stojanovski D et al (2022) Stimulating mitochondrial Biogenesis with deoxyribonucleosides increases functional capacity in ECHS1-Deficient cells. Int J Mol Sci 23:12610. https://doi.org/10.3390/ijms232012610
Burgin H, Sharpe AJ, Nie S et al (2023) Loss of mitochondrial fatty acid β-oxidation protein short‐chain Enoyl‐CoA hydratase disrupts oxidative phosphorylation protein complex stability and function. FEBS J 290:225–246. https://doi.org/10.1111/febs.16595
Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 33:W306-W310. https://doi.org/10.1093/nar/gki375
Carlston CM, Ferdinandusse S, Hobert JA et al (2019) Extrapolation of variant phase in mitochondrial short-chain Enoyl-CoA hydratase (ECHS1) Deficiency. In: Morava E, Baumgartner M, Patterson M et al (eds) JIMD reports, volume 43. Springer, Berlin, Heidelberg, pp 103–109
Chouchen J, Mahfood M, Alobathani M et al (2021) Clinical heterogeneity of the SLC26A4 gene in UAE patients with hearing loss and bioinformatics investigation of DFNB4/Pendred syndrome missense mutations. Int J Pediatr Otorhinolaryngol 140:110467. https://doi.org/10.1016/j.ijporl.2020.110467
D’Ordine RL, Tonge PJ, Carey PR, Anderson VE (1994) Electronic rearrangement induced by substrate analog binding to the enoyl-CoA hydratase active site: evidence for substrate activation. Biochemistry 33:12635–12643. https://doi.org/10.1021/bi00208a014
Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement - Haack −(2015) - Annals of Clinical and Translational Neurology - Wiley Online Library. https://onlinelibrary.wiley.com/doi/10.1002/acn3.189. Accessed 22 Aug 2023
Engel CK, Kiema TR, Hiltunen JK, Wierenga RK (1998) The Crystal structure of Enoyl-CoA hydratase complexed with Octanoyl-CoA reveals the structural adaptations required for binding of a long chain fatty Acid-CoA molecule. J Mol Biol 275:859–847
Ferdinandusse S, Waterham HR, Heales SJ et al (2013) HIBCH mutations can cause Leigh-like disease with combined deficiency of multiple mitochondrial respiratory chain enzymes and pyruvate dehydrogenase. Orphanet J Rare Dis 8:188. https://doi.org/10.1186/1750-1172-8-188
Ferdinandusse S, Friederich MW, Burlina A et al (2015) Clinical and biochemical characterization of four patients with mutations in ECHS1. Orphanet J Rare Dis 10:79. https://doi.org/10.1186/s13023-015-0290-1
Fitzsimons PE, Alston CL, Bonnen PE et al (2018) Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Am J Med Genet Part A 176:1115–1127. https://doi.org/10.1002/ajmg.a.38658
Fong JC, Schulz H (1977) Purification and properties of pig heart crotonase and the presence of short chain and long chain enoyl coenzyme A hydratases in pig and guinea pig tissues. J Biol Chem 252:542–547. https://doi.org/10.1016/S0021-9258(17)32751-5
Ganetzky RD, Bloom K, Ahrens-Nicklas R et al (2016) ECHS1 Deficiency as a cause of severe neonatal lactic acidosis. JIMD Rep 30:33–37. https://doi.org/10.1007/8904_2016_538
Hamed RB, Batchelar ET, Clifton IJ, Schofield CJ (2008) Mechanisms and structures of crotonase superfamily enzymes–how nature controls enolate and oxyanion reactivity. Cell Mol Life Sci 65:2507–2527. https://doi.org/10.1007/s00018-008-8082-6
Hass GM, Hill RL (1969) The subunit structure of crotonase. J Biol Chem 244:6080–6086
Lewin HA, Stewart-Haynes JA (1992) A simple method for DNA extraction from leukocytes for use in PCR. Biotechniques 13:522–524
Lim SC, Tajika M, Shimura M et al (2018) Loss of the mitochondrial fatty acid β-Oxidation protein medium-chain acyl-coenzyme A dehydrogenase disrupts oxidative phosphorylation protein Complex Stability and function. Sci Rep 8:153. https://doi.org/10.1038/s41598-017-18530-4
López-Ferrando V, Gazzo A, de la Cruz X et al (2017) PMut: a web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res 45:W222–W228. https://doi.org/10.1093/nar/gkx313
Matsushima N, Yoshida H, Kumaki Y et al (2008) Flexible structures and ligand interactions of tandem repeats consisting of proline, glycine, asparagine, serine, and/or threonine rich oligopeptides in proteins. Curr Protein Pept Sci 9:591–610. https://doi.org/10.2174/138920308786733886
Müller-Newen G, Janssen U, Stoffel W (1995) Enoyl-CoA hydratase and isomerase form a superfamily with a common active-site glutamate residue. Eur J Biochem 228:68–73. https://doi.org/10.1111/j.1432-1033.1995.tb20230.x
Nooren IMA, Thornton JM (2003) Diversity of protein-protein interactions. EMBO J 22:3486–3492. https://doi.org/10.1093/emboj/cdg359
Peters H, Buck N, Wanders R et al (2014) ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism. Brain 137:2903–2908. https://doi.org/10.1093/brain/awu216
Sahashi K, Masuda A, Matsuura T et al (2007) In vitro and in silico analysis reveals an efficient algorithm to predict the splicing consequences of mutations at the 5’ splice sites. Nucleic Acids Res 35:5995–6003. https://doi.org/10.1093/nar/gkm647
Sakai C, Yamaguchi S, Sasaki M et al (2015) ECHS1 mutations cause combined respiratory Chain Deficiency resulting in Leigh Syndrome. Hum Mutat 36:232–239. https://doi.org/10.1002/humu.22730
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–362. https://doi.org/10.1038/nmeth.2890
Sharpe AJ, McKenzie M (2018) Mitochondrial fatty acid oxidation disorders Associated with short-chain Enoyl-CoA hydratase (ECHS1) Deficiency. Cells 7:46. https://doi.org/10.3390/cells7060046
Simon MT, Eftekharian SS, Ferdinandusse S et al (2021) ECHS1 disease in two unrelated families of Samoan descent: common variant - rare disorder. Am J Med Genet Part A 185:157–167. https://doi.org/10.1002/ajmg.a.61936
Tetreault M, Fahiminiya S, Antonicka H et al (2015) Whole-exome sequencing identifies novel ECHS1 mutations in Leigh syndrome. Hum Genet 134:981–991. https://doi.org/10.1007/s00439-015-1577-y
Thirumal Kumar D, George Priya Doss C (2017) Role of E542 and E545 missense mutations of PIK3CA in breast cancer: a comparative computational approach. J Biomol Struct Dyn 35:2745–2757. https://doi.org/10.1080/07391102.2016.1231082
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Uchino S, Iida A, Sato A et al (2019) A novel compound heterozygous variant of ECHS1 identified in a Japanese patient with Leigh syndrome. Hum Genome Var 6:19. https://doi.org/10.1038/s41439-019-0050-1
Uesugi M, Mori J, Fukuhara S et al (2020) Short-chain enoyl-CoA hydratase deficiency causes prominent ketoacidosis with normal plasma lactate levels: a case report. Mol Genet Metabolism Rep 25:100672. https://doi.org/10.1016/j.ymgmr.2020.100672
Vaca Jacome AS, Rabilloud T, Schaeffer-Reiss C et al (2015) N-terminome analysis of the human mitochondrial proteome. Proteomics 15:2519–2524. https://doi.org/10.1002/pmic.201400617
Venegas V, Wang J, Dimmock D, Wong L-J (2011) Real-time quantitative PCR analysis of mitochondrial DNA content. Curr Protoc Hum Genet Chap 119:Unit 19.7. https://doi.org/10.1002/0471142905.hg1907s68
Wang M, Marín A (2006) Characterization and prediction of alternative splice sites. Gene 366:219–227. https://doi.org/10.1016/j.gene.2005.07.015
Yamada K, Aiba K, Kitaura Y et al (2015) Clinical, biochemical and metabolic characterisation of a mild form of human short-chain enoyl-CoA hydratase deficiency: significance of increased N-acetyl-S-(2-carboxypropyl)cysteine excretion. J Med Genet 52:691–698. https://doi.org/10.1136/jmedgenet-2015-103231
Yamashita A, Sugiura T, Waku K (1997) Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. J Biochem 122:1–16. https://doi.org/10.1093/oxfordjournals.jbchem.a021715
Yan BX, Sun YQ (1997) Glycine residues provide flexibility for enzyme active sites. J Biol Chem 272:3190–3194. https://doi.org/10.1074/jbc.272.6.3190
Yang H, Yu D (2020) Clinical, biochemical and metabolic characterization of patients with short-chain enoyl-CoA hydratase(ECHS1) deficiency: two case reports and the review of the literature. BMC Pediatr 20:50. https://doi.org/10.1186/s12887-020-1947-z
Yang Z, Cao J, Song Y et al (2022) Whole-exome sequencing identified novel variants in three Chinese Leigh syndrome pedigrees. Am J Med Genet Part A 188:1214–1225. https://doi.org/10.1002/ajmg.a.62641
Zhanhua C, Gan JG-K, Lei L et al (2005) Protein subunit interfaces: heterodimers versus homodimers. Bioinformation 1:28–39. https://doi.org/10.6026/97320630001028
Acknowledgements
We thank all the members of the family for their cooperation in the present study.
Funding
This work was supported by The Ministry of Higher Education and Scientifc Research in Tunisia and the University of Sharjah,UAE.
Author information
Authors and Affiliations
Contributions
MM: Elaborating the manuscript and conducting experimental analysis and interpretation of clinical, molecular, genetic, and in silico data. TK and LS: gathering, evaluating, and interpreting clinical data. Molecular and bioinformatics data analysis: OA, BK, MA, and RF. Overseeing the molecular genetic analyses are AT and CJ. MK: the manuscript has undergone a critical revision for intellectual content. FF: managing the research, collecting genetic information, and editing the manuscript critically for intellectual content. The final manuscript was read and approved by all authors.
Corresponding authors
Ethics declarations
Ethics approval
All procedures performed were in accordance with the ethical standards of the institutional research committee and with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Consent to participate
Written informed consent was obtained from the parents as legal guardians of the proband.
Consent for publication
We certifie that all contributors have read and approved the submission to this journal and that there is no financial or commercial involvement, or other conflict of interest by any author.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Maalej, M., Sfaihi, L., Fersi, OA. et al. Molecular and in silico investigation of a novel ECHS1 gene mutation in a consanguine family with short-chain enoyl-CoA hydratase deficiency and Mt-DNA depletion: effect on trimer assembly and catalytic activity. Metab Brain Dis 39, 611–623 (2024). https://doi.org/10.1007/s11011-024-01343-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-024-01343-6