
Abstract
Background
Mucopolysaccharidosis type IIIC (MPS IIIC; Sanfilippo syndrome C) is a rare lysosomal storage disease caused by mutations in the heparan-α-glucosaminide N-acetyltransferase (HGSNAT) gene, resulting in the accumulation of heparan sulfate. MPS IIIC is characterized by severe neuropsychiatric symptoms and mild somatic symptoms.
Methods
Our study analyzed the clinical presentation and biochemical characteristics of ten Chinese MPS IIIC patients from eight families. Whole exome sequencing was applied to identify the variants in HGSNAT gene. In one patient with only one mutant allele identified firstly, whole genome sequencing was applied. The pathogenic effect of novel variants was evaluated in silico.
Results
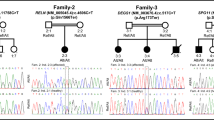
The mean age at the onset of clinical symptoms was 4.2 ± 2.5 years old, and the mean age of diagnosis was 7.6 ± 4.5 years old, indicating a delay of diagnosis. The most common onset symptoms were speech deterioration, and the most frequent presenting symptoms are speech deterioration, mental deterioration, hyperactivity and hepatomegaly, sequentially. All mutant alleles of 10 patients have been identified. There were eleven different HGSNAT variants, and the most common one was a previously reported variant c.493 + 1G > A. There were six novel variants, p.R124T, p.G290A, p.G426E, c.743 + 101_743 + 102delTT, c.851 + 171T > A and p.V582Yfs*18 in our cohort. Extraordinarily, two deep intron variants were identified in our cohort, with the variant c.851 + 171T > A identified by whole genome sequencing.
Conclusion
This study analyzed the clinical, biochemical, and genetic characteristics of ten Chinese MPS IIIC patients, which would assist in the early diagnosis and genetic counselling of MPS IIIC.




Similar content being viewed by others


Data Availability
The data for this study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Abbreviations
- MPS IIIC:
-
Mucopolysaccharidosis type IIIC
- HGSNAT:
-
heparan-α-glucosaminide N-acetyltransferase
- CPC:
-
cetylpyridinium chloride
- DMB:
-
1,9-dimethylmethene blue chloride
- GAGs:
-
glycosaminoglycans
- HS:
-
heparan sulfates
- DS:
-
dermatan sulfates
- CS:
-
Chondroitin sulfates
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
References
Andrade F, Aldamiz-Echevarria L, Llarena M, Couce ML (2015) Sanfilippo syndrome: overall review. Pediatr Int 57(3):331–338. https://doi.org/10.1111/ped.12636
Beneto N, Cozar M, Castilla-Vallmanya L, Zetterdahl OG, Sacultanu M, Segur-Bailach E, Garcia-Morant M, Ribes A, Ahlenius H, Grinberg D, Vilageliu L, Canals I (2020a) Neuronal and astrocytic differentiation from Sanfilippo C syndrome iPSCs for Disease modeling and Drug Development. J Clin Med 9(3). https://doi.org/10.3390/jcm9030644
Beneto N, Vilageliu L, Grinberg D, Canals I (2020b) Sanfilippo Syndrome: molecular basis, Disease Models and therapeutic approaches. Int J Mol Sci 21(21). https://doi.org/10.3390/ijms21217819
Berry HK (1987) Screening for mucopolysaccharide disorders with the berry spot test. Clin Biochem 20(5):365–371. https://doi.org/10.1016/s0009-9120(87)80088-7
Canals I, Beneto N, Cozar M, Vilageliu L, Grinberg D (2015) EXTL2 and EXTL3 inhibition with siRNAs as a promising substrate reduction therapy for Sanfilippo C syndrome. Sci Rep 5:13654. https://doi.org/10.1038/srep13654
Canals I, Elalaoui SC, Pineda M, Delgadillo V, Szlago M, Jaouad IC, Sefiani A, Chabas A, Coll MJ, Grinberg D, Vilageliu L (2011) Molecular analysis of Sanfilippo syndrome type C in Spain: seven novel HGSNAT mutations and characterization of the mutant alleles. Clin Genet 80(4):367–374. https://doi.org/10.1111/j.1399-0004.2010.01525.x
Chen X, Qiu W, Ye J, Han L, Gu X, Zhang H (2016) Demographic characteristics and distribution of lysosomal storage disorder subtypes in Eastern China. J Hum Genet 61(4):345–349. https://doi.org/10.1038/jhg.2015.155
de Jong JG, Wevers RA, Laarakkers C, Poorthuis BJ (1989) Dimethylmethylene blue-based spectrophotometry of glycosaminoglycans in untreated urine: a rapid screening procedure for mucopolysaccharidoses. Clin Chem 35(7):1472–1477. https://www.ncbi.nlm.nih.gov/pubmed/2503262
Fan X, Zhang H, Zhang S, Bagshaw RD, Tropak MB, Callahan JW, Mahuran DJ (2006) Identification of the gene encoding the enzyme deficient in Mucopolysaccharidosis IIIC (Sanfilippo Disease Type C). Am J Hum Genet 79(4):738–744. https://doi.org/10.1086/508068
Fedele AO (2015) Sanfilippo syndrome: causes, consequences, and treatments. Appl Clin Genet 8:269–281. https://doi.org/10.2147/TACG.S57672
Giugliani R, Federhen A, Michelin-Tirelli K, Riegel M, Burin M (2017) Relative frequency and estimated minimal frequency of lysosomal Storage Diseases in Brazil: Report from a Reference Laboratory. Genet Mol Biology 40(1):31–39. https://doi.org/10.1590/1678-4685-gmb-2016-0268
Griffin LS, Gloster TM (2017) The enzymatic degradation of Heparan Sulfate. Protein Pept Lett 24(8):710–722. https://doi.org/10.2174/0929866524666170724113452
Heron B, Mikaeloff Y, Froissart R, Caridade G, Maire I, Caillaud C, Levade T, Chabrol B, Feillet F, Ogier H, Valayannopoulos V, Michelakakis H, Zafeiriou D, Lavery L, Wraith E, Danos O, Heard JM, Tardieu M (2011) Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A 155A(1):58–68. https://doi.org/10.1002/ajmg.a.33779
Hopwood JJ, Harrison JR (1982) High-resolution electrophoresis of urinary glycosaminoglycans: an improved screening test for the mucopolysaccharidoses. Anal Biochem 119(1):120–127. https://doi.org/10.1016/0003-2697(82)90674-1
Hřebíček M, Mrázová L, Seyrantepe V, Durand S, Roslin NM, Nosková L, Hartmannová H, Ivánek R, Čížková A, Poupětová H, Sikora J, Uřinovská J, Stránecký V, Zeman J, Lepage P, Roquis D, Verner A, Ausseil J, Beesley CE, Maire I, Poorthuis BJHM, Kamp Jvd D, OPv, Wevers RA, Hudson TJ, Fujiwara TM, Majewski J, Morgan K, Kmoch S, Pshezhetsky AV (2006) Mutations in TMEM76* cause Mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am J Hum Genet 79(5):807–819. https://doi.org/10.1086/508294
Hult M, Darin N, Döbeln Uv, Månsson J-E (2014) Epidemiology of lysosomal storage diseases in Sweden. Acta Paediatr 103(12):1258–1263. https://doi.org/10.1111/apa.12807
Kim MS, Yang A, Noh ES, Kim C, Bae GY, Lim HH, Park HD, Cho SY, Jin DK (2022) Natural history and molecular characteristics of korean patients with mucopolysaccharidosis type III. J Pers Med 12(5). https://doi.org/10.3390/jpm12050665
Klein U, Kresse H, Figura Kv (1978). Sanfilippo syndrome type C: deficiency of acetyl-CoA:alpha-glucosaminide N-acetyltransferase in skin fibroblasts. Proceedings of the National Academy of Sciences, 75(10), 5185–5189. https://doi.org/10.1073/pnas.75.10.5185
Kong W, Meng Y, Zou L, Yang G, Wang J, Shi X (2020a) Mucopolysaccharidosis III in Mainland China: natural history, clinical and molecular characteristics of 34 patients. J Pediatr Endocrinol Metab 33(6):793–802. https://doi.org/10.1515/jpem-2019-0505
Kong W, Yao Y, Zhang J, Lu C, Ding Y, Meng Y (2020b) Update of treatment for mucopolysaccharidosis type III (sanfilippo syndrome). Eur J Pharmacol 888:173562. https://doi.org/10.1016/j.ejphar.2020b.173562
Kurihara M, Kumagai K, Yagishita S (1996) Sanfilippo syndrome type C: a clinicopathological autopsy study of a long-term survivor. Pediatr Neurol 14(4):317–321. https://doi.org/10.1016/0887-8994(96)00083-5
Lin HY, Chuang CK, Lee CL, Tu RY, Lo YT, Chiu PC, Niu DM, Fang YY, Chen TL, Tsai FJ, Hwu WL, Lin SJ, Chang TM, Lin SP (2018) Mucopolysaccharidosis III in Taiwan: natural history, clinical and molecular characteristics of 28 patients diagnosed during a 21-year period. Am J Med Genet A 176(9):1799–1809. https://doi.org/10.1002/ajmg.a.40351
Martins C, de Medeiros PFV, Leistner-Segal S, Dridi L, Elcioglu N, Wood J, Behnam M, Noyan B, Lacerda L, Geraghty MT, Labuda D, Giugliani R, Pshezhetsky AV (2019) Molecular characterization of a large group of mucopolysaccharidosis type IIIC patients reveals the evolutionary history of the disease. Hum Mutat 40(8):1084–1100. https://doi.org/10.1002/humu.23752
Meikle PJ (1999) Prevalence of lysosomal Storage Disorders. JAMA 281(3):249. https://doi.org/10.1001/jama.281.3.249
Meyer A, Kossow K, Gal A, Muhlhausen C, Ullrich K, Braulke T, Muschol N (2007) Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics 120(5):e1255–1261. https://doi.org/10.1542/peds.2007-0282
Minoche AE, Lundie B, Peters GB, Ohnesorg T, Pinese M, Thomas DM, Zankl A, Roscioli T, Schonrock N, Kummerfeld S, Burnett L, Dinger ME, Cowley MJ (2021) ClinSV: clinical grade structural and copy number variant detection from whole genome sequencing data. Genome Med 13(1):32. https://doi.org/10.1186/s13073-021-00841-x
Neufeld EF, Muenzer J (2019) The Mucopolysaccharidoses. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA (eds) The online metabolic and molecular bases of inherited disease. McGraw-Hill Education. ommbid.mhmedical.com/content.aspx?aid=1181463720
Ouesleti S, Coutinho MF, Ribeiro I, Miled A, Mosbahi DS, Alves S (2017) Update of the spectrum of mucopolysaccharidoses type III in Tunisia: identification of three novel mutations and in silico structural analysis of the missense mutations. World J Pediatr 13(4):374–380. https://doi.org/10.1007/s12519-017-0005-x
Pennock CA (1976) A review and selection of simple laboratory methods used for the study of glycosaminoglycan excretion and the diagnosis of the mucopolysaccharidoses. J Clin Pathol 29(2):111–123. https://doi.org/10.1136/jcp.29.2.111
Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H, Pinto E, Silva E, Rocha S, Marcao A, Ribeiro I, Lacerda L, Ribeiro G, Amaral O, Sa Miranda MC (2004) Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet 12(2):87–92. https://doi.org/10.1038/sj.ejhg.5201044
Poorthuis BJHM, Wevers RA, Kleijer WJ, Groener JEM, Jong JGNd W, Sv, Niezen-Koning KE, Diggelen OPv (1999) The frequency of lysosomal storage diseases in the Netherlands. Hum Genet 105(1–2):151–156. https://doi.org/10.1007/s004399900075
Poupetova H, Ledvinova J, Berna L, Dvorakova L, Kozich V, Elleder M (2010) The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 33(4):387–396. https://doi.org/10.1007/s10545-010-9093-7
Ruijter GJ, Valstar MJ, van de Kamp JM, van der Helm RM, Durand S, van Diggelen OP, Wevers RA, Poorthuis BJ, Pshezhetsky AV, Wijburg FA (2008) Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in the Netherlands. Mol Genet Metab 93(2):104–111. https://doi.org/10.1016/j.ymgme.2007.09.011
Schiff ER, Daich Varela M, Robson AG, Pierpoint K, Ba-Abbad R, Nutan S, Zein WM, Ullah E, Huryn LA, Tuupanen S, Mahroo OA, Michaelides M, Burke D, Harvey K, Arno G, Hufnagel RB, Webster AR (2020) A genetic and clinical study of individuals with nonsyndromic retinopathy consequent upon sequence variants in HGSNAT, the gene associated with Sanfilippo C mucopolysaccharidosis. Am J Med Genet C Semin Med Genet 184(3):631–643. https://doi.org/10.1002/ajmg.c.31822
Sun Y, Peng J, Liang D, Ye X, Xu N, Chen L, Yan D, Zhang H, Xiao B, Qiu W, Shen Y, Pang N, Liu Y, Liang C, Qin Z, Luo J, Chen F, Wang J, Zhang Z, Wei H, Du J, Yan H, Duan R, Wang J, Zhang Y, Liao S, Sun K, Wu L, Yu Y (2022) Genome sequencing demonstrates high diagnostic yield in children with undiagnosed global developmental delay/intellectual disability: a prospective study. Hum Mutat 43(5):568–581. https://doi.org/10.1002/humu.24347
Sun Y, Ye X, Fan Y, Wang L, Luo X, Liu H, Gao X, Gong Z, Wang Y, Qiu W, Zhang H, Han L, Liang L, Ye H, Xiao B, Gu X, Yu Y (2020) High detection rate of Copy Number Variations using capture sequencing data: a retrospective study. Clin Chem 66(3):455–462. https://doi.org/10.1093/clinchem/hvz033
Tordo J, O’Leary C, Antunes A, Palomar N, Aldrin-Kirk P, Basche M, Bennett A, D’Souza Z, Gleitz H, Godwin A, Holley RJ, Parker H, Liao AY, Rouse P, Youshani AS, Dridi L, Martins C, Levade T, Stacey KB, Davis DM, Dyer A, Clement N, Bjorklund T, Ali RR, Agbandje-McKenna M, Rahim AA, Pshezhetsky A, Waddington SN, Linden RM, Bigger BW, Henckaerts E (2018) A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain 141(7):2014–2031. https://doi.org/10.1093/brain/awy126
Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA (2008) Sanfilippo syndrome: a mini-review. J Inherit Metab Dis 31(2):240–252. https://doi.org/10.1007/s10545-008-0838-5
Voznyi YV, Karpova EA, Dudukina TV, Tsvetkova IV, Boer AM, Janse HC, van Diggelen OP (1993) A fluorimetric enzyme assay for the diagnosis of Sanfilippo disease C (MPS III C) Journal of inherited metabolic disease(2)
Acknowledgements
We are greatly indebted to the patients and their families who participated in this study for their invaluable contributions, which made the work described in this manuscript possible.
Funding
This work was supported by the Shanghai Municipal Health Commission [grant number 202240361]; the Shanghai Science & Technology Commission [grant number 20ZR1446300]; and the Shanghai Municipal Education Commission [grant number 20152520]. These funding did not have any role in the conduct of the research, preparation of the article or the decision to submit the article for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Consent to participate
Written informed consent was obtained from the guardians of children.
Consent to publication
Written informed consent was obtained from the guardians of children.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liang, Y., Gao, X., Lu, D. et al. Mucopolysaccharidosis type IIIC in chinese mainland: clinical and molecular characteristics of ten patients and report of six novel variants in the HGSNAT gene. Metab Brain Dis 38, 2013–2023 (2023). https://doi.org/10.1007/s11011-023-01204-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-023-01204-8