
Abstract
Patients with liver disease have reduced urea synthesis capacity resulting in reduced capacity to detoxify ammonia in the liver. The contribution of the gut to the hyperammonemic state observed during liver failure is mainly due to portacaval shunting and not the result of changes in the metabolism of ammonia in the gut. Small intestinal synthesis of ammonia is related to amino acid breakdown, predominantly glutamine, whereas large bowel ammonia production is caused by bacterial breakdown of amino acids and urea. The kidneys produce ammonia but adapt to liver failure in experimental portacaval shunting by reducing ammonia release into the systemic circulation. The kidneys have the ability to switch from net ammonia production to net ammonia excretion. Data from recent studies in patients with cirrhosis of the liver show that the kidneys have a major role in post upper gastrointestinal bleeding hyperammonemia. During hyperammonemia muscle takes up ammonia and plays a major role in (temporarily) detoxifying ammonia to glutamine. Net uptake of ammonia by the brain occurs in patients and experimental animals with acute and chronic liver failure. Insight will be given in recent developments on ammonia lowering therapies which are based on the information of interorgan ammonia trafficking.
Similar content being viewed by others


General introduction
The liver has a key function in nitrogen metabolism. Liver disease and the resulting hepatocellular failure adversely affect these processes and induce profound disturbances in amongst others nitrogen homeostasis. A crucial feature in this context is the diminished hepatic urea synthesis capacity, leading to an impaired capacity to detoxify ammonia. Glutamine synthesis has been suggested to be the most important alternative pathway for ammonia detoxification during diminished urea synthesis. In the past 15 years, we have focused on interorgan exchange of amino acids and ammonia during acute and chronic liver failure and in this review we will address this issue and pay special emphasis on the effects of upper gastrointestinal bleeding on it.
Ammonia and glutamine
Although ammonia is important in several biochemical pathways, it is toxic at elevated levels (Zieve 1987, Cooper and Plum 1987). Therefore, it must be converted to a non-toxic compound and several pathways serve this purpose. In most mammals, this takes place mainly in the liver by synthesis of urea in the urea-cycle (ornithine cycle) (Cooper and Plum 1987;Meijer et al. 1990). During liver failure diminished urea synthesis capacity and portasystemic shunting (Zieve 1987; Meijer et al. 1990) impairs or bypasses the main route of ammonia detoxification (urea synthesis), leading to hyperammonemia. In this situation the synthesis of glutamine (see below) from equimolar amounts of glutamate and ammonia becomes the most important, though temporary, pathway for ammonia detoxification (Meijer et al. 1990).
Glutamine is a non-toxic, non-essential amino acid, consisting of a five-carbon chain and two nitrogen residues. Of all amino acids that can be incorporated in protein, glutamine is the most abundant: it has the highest plasma concentration, and constitutes approximately 50% of the whole body free amino acid pool (Lacey and Wilmore 1990). Glutamine serves as an obligatory fuel for intestinal and other rapidly dividing cells e.g. the immune system (Lacey and Wilmore 1990) and plays an important role in the regulation of acid/base balance by providing the most important substrate for renal ammoniagenesis in many mammals (Halperin et al. 1992).
Ammonia and glutamine exchange across the gut
In the physiological state, glutamine is a crucial source of energy for the large and especially small intestine. The intestines take up glutamine in large quantities from either the blood-stream or the intestinal lumen (Windmueller 1982). Most of the glutamine consumed by the intestines is utilized in the small intestinal mucosa (predominantly jejunum), constituting 75% of small intestinal weight.
The initial step in intestinal glutamine breakdown is by conversion of glutamine to glutamate and ammonia, a reaction catalysed by the enzyme glutaminase. The intestines contain high glutaminase activity and only minimal glutamine synthetase activity, which makes this organ particularly suitable to use glutamine as an energy source. On the other hand, it should be stressed that ammonia is a toxic product of this reaction. The large bowel utilizes less glutamine, but instead uses other substrates e.g. glucose, short chain fatty acids and ketone bodies (Weber and Veach 1979). Apart from this small contribution to glutamine metabolism–derived ammonia, the colon contributes significantly to portal venous ammonia generation by bacterial splitting of urea and possibly amino acids (Weber and Veach 1979).
Intestinal ammonia and glutamine metabolism during liver failure
Intestinal ammonia production in portacaval shunted rats is similar to healthy post-absorptive rats (400 nmol/100 g bw/min), and is in stochiometric proportion with glutamine consumption (Souba et al. 1990). During acute liver ischemia in rats there is an initial small increase of intestinal ammonia production and glutamine consumption. However, 4 h after the induction of liver ischemia, glutamine consumption ceased whereas ammonia production remained approximately 300 nmol/100 g bw/min despite an increase in arterial glutamine concentration from about 800 µM to 2300 µM (Dejong et al. 1992). These observations are in agreement with the in vitro data which suggest that intestinal glutamine uptake increases linearly to a plateau at arterial concentrations of 600 µM.
For obvious reasons, studies on intestinal ammonia and glutamine metabolism in patients with liver disease are sparse. Webster and colleagues (Webster et al. 1958) studied changes in ammonia concentrations in superficial abdominal veins, representing portal blood. They showed increased ammonia concentrations after oral administration of protein and glutamine, but did not relate this to changes in arterial ammonia levels. Owen and colleagues showed uptake of glutamine by the portal drained viscera via selective umbilicoportal cannulation in patients with cirrhosis of the liver with (Owen et al. 1985) and without portasystemic shunts (Owen et al. 1981). However, they did not measure ammonia concentrations. Recently, Plauth and colleagues reported negative arterio-venous concentration differences of ammonia across the superior mesenteric vein in overnight fasted patients with cirrhosis of the liver that where treated with a TIPSS (Plauth et al. 2000). It is difficult to interpret their data because the patients they studied consisted of both stable post-absorptive cirrhotics with a TIPSS and patients who were undergoing portosystemic shunting for recurrent variceal bleeding. Moreover, the patients received an oral decontamination protocol to reduce ammonia production from intestinal bacteria. We recently studied the quantitative dynamics of interorgan ammonia and amino acid metabolism in 24 metabolically stable patients with cirrhosis who underwent TIPSS portography (Olde Damink et al. 2002a). Blood was sampled and blood flows measured across portal drained viscera, leg, kidney and liver, and arterio-venous differences across the spleen and the inferior and superior mesenteric veins. We found that ammonia was predominantly produced by the portal drained viscera. Glutamine was the only amino acid that was taken up by the portal drained viscera. PDV uptake of glutamine was significantly correlated with PDV production of ammonia (r = −0.809, p < 0.001), citrulline (r = −0.863, p < 0.001) and weakly with production of alanine (r = 0.444, p = 0.034). The contribution of the intestines to hyperammonemia in chronic liver failure was the result of existing intra–and/or extrahepatic portacaval shunts and not of altered intestinal ammonia production.
The association between high protein loads, hyperammonemia and hepatic encephalopathy in patients with hepatic failure has led to the generally accepted idea that enhanced intestinal ammonia liberation is an important factor in the pathogenesis of hepatic encephalopathy (Zieve 1987; Olde Damink et al. 1999). However, this could not be proven in our study in patients with cirrhosis of the liver in whom a upper GI bleed was simulated by administration of an amino acid solution mimicking haemoglobin (Olde Damink et al. 2003). During the simulated bleed arterial concentrations of ammonia increased significantly. There was no change in ammonia production from the portal drained viscera but renal ammonia production increased 6-fold. In the second part of this study we studied 9 cirrhotic patients with a real upper GI bleed that underwent acute TIPSS insertion for treatment of their bleed (Olde Damink et al. 2003). We could clearly demonstrate that ammonia was only produced by the kidneys and not by the splanchnic area, indicating the pivotal role of the kidney in ammonia homeostasis (see Fig. 1 and 2).

The role of the kidney in inter-organ glutamine (GLN) exchange under physiological circumstances. GLN is taken up by the gut and kidney and metabolised by the glutaminase (G-ase) pathway to yield ammonia (AMM). AMM generated by the kidney is excreted in the urine or released into the renal vein. Renal AMM released into the circulation as well as AMM generated by the gut is metabolised in the liver in periportal hepatocytes to form urea, the latter being excreted in the urine (the role of muscle and brain in GLN synthesis has been omitted deliberately from the graph)

The proposed role of the kidney in inter-organ glutamine exchange during hyperammonemia due to liver failure (cf. also Fig. 1). Urea synthesis is diminished and AMM coming from the gut or kidneys escapes periportal urea synthesis. This AMM is subsequently scavenged in the perivenous hepatocytes in the glutamine synthetase pathway (GS) to form GLN. GLN in turn is released back into the circulation and subsequently undergoes degradation by G-ase in gut and kidney. During hyperammonemia, a greater fraction of the AMM generated in the kidney is excreted in the urine, which turns the kidney into an organ of net ammonia removal from the body (the role of muscle and brain in GLN synthesis has been omitted deliberately from the graph)
Ammonia and glutamine exchange across the liver
Ammonia and amino acids produced by the gut are released into the portal blood and transported to the liver. Ammonia detoxification in the liver occurs mainly in periportal hepatocytes in the urea cycle (ornithine cycle), leading to urea formation. Urea is subsequently released into the hepatic veins and then excreted in the urine. Hepatic urea synthesis is a low affinity, high capacity detoxification system (Meijer et al. 1990); (Haussinger et al. 1988) and normally very little ammonia escapes this process. Any ammonia escaping detoxification in the periportal hepatocytes is usually trapped in the perivenous hepatocytes, where it is incorporated in glutamine in the glutamine synthetase reaction (a high affinity, low capacity system) (Meijer et al. 1990); (Haussinger et al. 1988).
Hepatic ammonia, urea and glutamine metabolism are compartmentalized in the liver. The liver contains both glutaminase and glutamine synthetase activity, but these enzymes have a zonated distribution pattern in the liver (Haussinger et al. 1988). Glutamine synthetase activity is restricted to a small hepatocyte population immediately surrounding the hepatic venules, whereas glutaminase activity is mainly located in the periportal zone. Urea cycle activity is localized in the periportal hepatocytes. It is of particular interest to note that glutaminase in periportal hepatocytes has the extraordinary characteristic of being activated by its product ammonia (Haussinger et al. 1988), as opposed to most other types of glutaminase, that are inhibited by ammonia.
Liver ammonia and glutamine metabolism during liver failure
Patients with cirrhosis of the liver have intra– and/or extrahepatic portasystemic shunts that can account for up to 50% of the portal blood flow. This may be even higher in patients with iatrogenically created portosystemic shunts (93%) (Walser et al. 1996). Furthermore, patients with cirrhosis have reduced ammonia detoxification capacity because of diminished urea and glutamine synthesis capacity. In these patients the flux through glutaminase is 4 to 6 fold increased (Kaiser et al. 1988). This has been interpreted as a compensatory mechanism to maintain a normal flux through the urea cycle despite the decrease in urea cycle enzyme activities. However, peri-venous glutamine synthetase activity is not correspondingly increased and ammonia ions escape clearance by the liver and are released into the systemic circulation (Haussinger 1986).
The above mentioned mechanism can explain the high hepatic venous ammonia concentrations observed in post-absorptive patients with chronic liver disease (40–60 μM), acute-on-chronic liver disease (70–90 μM), and acute liver failure (200–240 μM) (Clemmesen et al. 1999, 2000). There was a high correlation between arterial and hepatic venous ammonia concentrations (r = 0.85) (Clemmesen et al. 1999). Ammonia production across the hepato-splanchnic area did not differ from zero in patients with chronic and acute-on-chronic liver disease, suggesting that the liver was still able to remove all the ammonia that was delivered to it from the portal vein and the hepatic artery. The patients with acute liver failure showed net ammonia release from the hepato-splanchnic region (110 mmol/min), suggesting defective ammonia scavenging by the liver. This was accompanied by net glutamine uptake (240 mmol/min). The observed hepato-splanchnic ammonia release does not exclude hepatic ammonia removal because there was no information about portal venous ammonia concentrations. Recently, we studied hepatic ammonia, glutamine and urea exchange in 24 patients with stable hepatic cirrhosis and previous treatment with a transjugular intrahepatic portasystemic stent shunt (TIPSS) undergoing TIPSS portography to check shunt patency (Olde Damink et al. 2002b). The cirrhotic liver still removed ammonia from the circulation in significant quantities. In addition, glutamine and alanine uptake from the portal blood stream accounted for 50% of total amino acid uptake by the liver (Olde Damink et al. 2002b). Hepatic urea synthesis was significantly correlated with glutamine and alanine uptake. Induction of hyperammonemia by administration of a simulated upper GI bleed did not result in enhanced ammonia uptake by the liver, suggesting a maximum plateau (Olde Damink et al. 1999).
Ammmonia and glutamine exchange across muscle
In a situation where the liver fails to clear the ammonia generated within the splanchnic bed, other organs will be forced to adapt to a situation of high systemic ammonia levels. Glutamine synthesis is the most important alternative detoxification pathway in this situation and muscle and brain have traditionally been proposed to play a key role in this.
Glutamine synthetase activity in skeletal muscle is low (Lund 1970), but by virtue of its mass, muscle is one of the principal glutamine synthesizing organs. Skeletal muscle glutaminase activity (Nelson et al. 1992) is negligible compared to glutamine synthetase activity and therefore, muscle should be viewed as a glutamine synthesizing organ. Ammonia can be taken up or released by skeletal muscle. Ammonia uptake by skeletal muscle was shown in various hyperammonemic states in rats, dogs and man, but also in healthy human volunteers and normal control rats (reviewed in detail in (Olde Damink et al. 2002a)). Unidirectional ammonia uptake by skeletal muscle has also been shown in patients with various degrees of liver failure (Lockwood et al. 1979). Data from arterio-venous catheterization studies suggest that net ammonia uptake by muscle is probably related to (changes in) arterial ammonia levels. Net muscle ammonia uptake was shown in hyperammonemic patients with (decompensated) cirrhosis of the liver in arterio-venous difference measurements across the forearm and leg (reviewed in detail in (Olde Damink et al. 2002a)). A good correlation between arterial ammonia concentrations and muscle ammonia uptake was observed in patients in hepatic coma with (Webster and Gabuzda 1958) or without (Bessman and Bradley, 1955) active upper gastrointestinal hemorrhage and in patients with decompensated cirrhosis (Tyor et al. 1960) and gross muscle wasting (Ganda and Ruderman 1976). No significant correlation was found in stable patients with cirrhosis of the liver (Webster and Gabuzda 1958) with normal muscle composition (Ganda and Ruderman 1976). The fractional extraction of ammonia was reduced in patients with muscle wasting (Ganda and Ruderman 1976), suggesting an important role for maintaining adequate muscle mass in these patients. Arterio-venous differences do not reflect the rate of production or consumption of ammonia since the latter also depends on organ flow. Organ flux measurements are necessary to enable quantification of metabolism and comparison of individual organs. Recently, net ammonia consumption by leg muscle has been quantified in patients with acute liver failure (Clemmesen et al. 2000) and was calculated to be 100 nmol/100 g/min. The authors did not document the correlation between arterial ammonia concentration and ammonia consumption by muscle, but leg ammonia consumption was correlated with glutamine production supporting the view that skeletal muscle participates in ammonia detoxification in acute liver failure. Total amino acid production from muscle was 10 times higher than uptake of ammonia, indicating that production of glutamine was not only the result of ammonia consumption but also the result of net muscle catabolism during acute liver failure (Clemmesen et al. 2000). Recent data from our interorgan ammonia metabolism study demonstrated an important role for muscle in ammonia removal in metabolically stable patients with cirrhosis of the liver and a TIPSS in situ (Olde Damink et al. 2002b). Significant ammonia uptake by the leg was observed and glutamine release in this study exceeded ammonia uptake. Induction of hyperammonemia by administration of a simulated upper GI bleed resulted in a significant increase in muscle ammonia, in contrast to an unchanged ammonia removal by the liver (Olde Damink et al. 2003). Ammonia uptake by muscle, leading to glutamine synthesis and glutamine release from muscle does not necessarily indicate net ammonia detoxification. Glutamine released from skeletal muscle can be taken up in the splanchnic area or in the kidneys and can be metabolized to ammonia, abolishing the effect on whole body ammonia removal (Olde Damink et al. 2003). Glutamine acts thereby as a non-toxic nitrogen carrier, without contributing to net nitrogen removal from the systemic circulation.
In conclusion, skeletal muscle metabolism probably plays an important role in ammonia metabolism in chronic and acute liver failure. This is mainly because total body skeletal muscle mass probably constitutes the quantitatively most important localization of the enzyme glutamine synthestase.
Ammonia and glutamine exchange across the brain
The brain has traditionally been thought to play a crucial role in glutamine synthesis during hyperammonemia due to liver failure. Brain contains appreciable amounts of both glutamine synthetase and glutaminase (Cooper and Plum 1987). These two enzymes are compartmentalized in brain: astrocytes contain most of total brain glutamine synthetase whilst neurons contain virtually all brain glutaminase (Cooper and Plum 1987). The normal brain is generally viewed as an organ of ammonia uptake and glutamine release. Ammonia uptake has been suggested to occur in healthy human volunteers (Warter et al. 1983a, 1983b) and in some animal species (Cooper and Plum 1987). Tracer studies using 13N-ammonia have demonstrated that in the normal brain ammonia is rapidly incorporated in glutamine (t ½ < 3 sec) (Cooperet al. 1979), suggesting that cerebral ammonia detoxification is by the glutamine synthetase reaction (Cooper and Plum 1987; Cooper et al. 1979). This takes place in a small (20% volume) compartment of the brain, probably the astrocytes (Cooper and Plum 1987); (Cooper et al. 1979). During hyperammonemia, ammonia is less rapidly incorporated in glutamine (t ½ = 9 sec).
Net cerebral ammonia uptake during chronic hyperammonemia has been demonstrated in humans (Clemmesen et al. 1999; Warter et al. 1983a, 1983b; Jalan et al. 1999). Although never tested in comparative study, it can be hypothesized that brain glutamine synthetic capacity is negligible compared with that of skeletal muscle, in view of their respective relative masses.
Ammonia and glutamine exchange across the kidney
The kidneys play a pivotal role in waste nitrogen excretion. Also, renal glutamine and ammonia metabolism plays an important role in acid-base regulation (Halperin et al. 1992). With respect to both nitrogen excretion and acid-base equilibrium, the kidneys and liver interact in a very sophisticated manner to maintain whole body homeostasis. It is therefore surprising that renal ammonia and glutamine metabolism have received relatively little attention in patients with liver disease.
The kidneys contain both glutaminase and glutamine synthetase (Welbourne et al. 1986) and therefore, the kidneys are capable of both synthesizing and degrading glutamine. In the postabsorptive state, glutamine is taken up by the mammalian kidney (Welbourne et al. 1986). Glutamine constitutes the main substrate for renal ammoniagenesis (Halperin et al. 1992). Following uptake from the bloodstream, it is metabolised by intra-mitochondrial phosphate-dependent glutaminase and the ammonia produced is either excreted in the urine or released back into the renal vein (Halperin et al. 1992) (see Fig. 1).
In the physiological situation, the mammalian kidneys excrete only minor amounts of ammonia in the urine: 30% of total renal ammonia production is released into the urine, the remainder is released into the renal vein (Halperin et al. 1992) (Fig. 1). These figures illustrate that the normal kidney is an organ of net ammonia addition to the body. During acidosis, this situation reverses because total renal ammoniagenesis is enhanced and 70% of this enhanced amount is excreted in the urine to dispose the acid load (Halperin et al. 1992). Glutamine breakdown and subsequent ammonia excretion in the urine are a means of excreting protons and generating bicarbonate (Halperin et al. 1992). Bicarbonate is used in hepatic urea synthesis, which therefore also constitutes a pH-regulating modality (Meijer et al. 1990). This illustrates the sophisticated way in which liver and kidney act in concert to maintain nitrogen and acid-base homeostasis.
In the past, high glutamine and ammonia concentrations were suggested to favor urinary ammonia excretion (Tyor et al. 1960); (Owen et al. 1961) and it was shown that the kidney releases ammonia into the circulation in cirrhotic patients (Tyor et al. 1960). This renal ammonia release into the circulation decreased at elevated ammonia concentrations (Tyor et al. 1960). Similarly, artificially elevated arterial ammonia levels in human volunteers induced the kidney to take up ammonia from the circulation and to increase urinary ammonia excretion (Owen et al. 1961). A problem of these studies is that most of them do not provide data on renal glutamine metabolism.
In the early nineties of the past century, we have conducted two rat experiments during acute (Dejong et al. 1993a) and chronic (Dejong et al. 1993b) hyperammonemia. In these experiments, 70% of total renal ammoniagenesis was excreted in the urine and only 30% was released back into the renal vein. This reversal of the urinary excretion/renal venous release ratio from 30/70 to 70/30, made the kidneys change from ammonia-producing organs in the body to ammonia elimination. This beneficial adaptation helps lowering systemic ammonia levels (see Fig. 2).
Subsequent experiments during feeding in pigs have shown that renal ammoniagenesis increases following a protein meal and that the kidneys contribute more to systemic ammonia levels than the entire hepatosplanchnic area (Welters et al. 1999). These results have recently been confirmed in patients with hepatic cirrhosis studied under control conditions (Olde Damink et al. 2002b) and during an actual as well as a simulated gastrointestinal bleed (Olde Damink et al. 2003). During the simulated bleed arterial concentrations of ammonia increased significantly (p = 0.002). There was no change in ammonia production from the portal drained viscera but renal ammonia production increased 6-fold (p = 0.008). In contrast to an unchanged ammonia removal by the liver, a significant increase in muscle ammonia removal was observed. In patients with an acute UGI bleed, ammonia was only produced by the kidneys (572 (184) nmol/kgbw/min) and not by the splanchnic area (−121 (87) nmol/kgbw/min) (Olde Damink et al. 2003).
Other organs
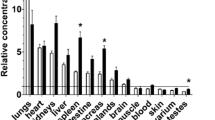
In the past, some attention has also been paid to the role of the lung in glutamine and ammonia homeostasis (Welbourne 1988; Plumley et al. 1990). Some authors have suggested that the normal (Welbourne 1988) as well as injured (Plumley et al. 1990) lung is an organ of glutamine release. Incidentally, ammonia uptake by the lung in septic patients has been observed (Plumley et al. 1990). The enzymatic characteristics of lung tissue (lungs contain glutamine synthetase) suggests a role for the lungs in ammonia uptake and glutamine release during liver failure. This has recently been proven in a study in pigs with acute liver failure (Ytrebø et al. 2006). Literature concerning the role of the heart in ammonia and glutamine exchange is scarce. Considering the enzymatic properties of myocardial cells (Lund 1970; Nelson et al. 1992), the heart could contribute to glutamine production, as well as breakdown. To our knowledge, no data are available concerning ammonia and glutamine metabolism by the heart during liver failure.
New insight in ammonia lowering strategies during hyperammonemia
As detailed above, we have generated data in the past 15 years clearly showing that the kidney may play a crucial role in ammonia metabolism during acute and chronic liver failure by changing from ammonia production in, to ammonia excretion from the body. These results have recently been confirmed in patients with hepatic cirrhosis studied under control conditions (Olde Damink et al. 2002b) and during an actual as well as a simulated gastrointestinal bleed (Olde Damink et al. 2003). Taken together, these animal and human studies constitute a considerable and consistent body of evidence that provide us with a basis for suggesting two important modifications of the prevailing ammonia hypothesis of hepatic encephalopathy:
-
fasted systemic ammonia levels during liver disease are probably determined by intestinal and renal ammonia metabolism
-
hyperammonemia following an intestinal protein load (meal, gastrointestinal bleed) is not only the consequence of intestinal ammonia generation, but is predominatly caused by renal ammonia generation
Clearly, this modified hypothesis provides us with a unique opportunity to start a search for new ammonia-lowering therapeutic modalities and help explain why the traditional therapeutic modalities that solely focussed on intestinal ammonia production had limited clinical effect. These treatments aim at accelerating intestinal transit time by cathartic agents, reducing protein loads, lowering systemic ammonia levels by acidification of intestinal contents with lactulose and reducing bacterial flora with poorly resorbable antibiotics. The most likely candidate for therapeutic intervention is reduction of renal ammoniagenesis, but to the best of our knowledge no clinical advances are made to modify renal ammonia excretion.
Recently a interesting concept of ammonia detoxification was developed in one of our labs that combines the (temporary) ammonia buffering of l-ornithine with the renal excretion of glutamine bound phenylacetate (detailed in (Jalan et al. 2007)). The concept of delivery of l-ornithine to the muscle is the basis of the agent l-ornithine l-aspartate for ammonia lowering in cirrhosis. l-ornithine l-aspartate provides l-ornithine and l-aspartate as substrates for glutamate production. Randomized controlled trials (RCT’s) of l-ornithine l-aspartate administration to patients with chronic liver failure, hyperammonemia and subclinical HE, have reported reductions in plasma ammonia and improved psychometric test scores (Staedt et al. 1993) and (Kircheis et al. 1997). However, the results of the use of l-ornithine l-aspartate are not universally positive, with a number of apparently negative trials yet to be published.
Phenylbutyrate (converted to phenylacetate in vivo) has been used for the hyperammonemia which occurs with urea cycle enzyme deficiencies (Smith 1981). Phenylacetate covalently combines with the glutamine derived from glutamate to make phenylacetylglutamine which is excreted by the kidneys. In such metabolic disorders elevated glutamine levels act as the substrate for phenylacetate, thereby removing it as a substrate for ammoniagenesis.
The above studies of inter-organ ammonia trafficking and the lessons from l-ornithine l-aspartate observations and the current use of phenylacetate (often administered as phenylbutyrate) to treat urea cycle enzyme disorders have led up to the hypothesis: the concomitant administration of l-ornithine and phenylbutyrate or phenylacetate in liver failure produces a sustained reduction in ammonia concentration through: 1. provision of glutamate (by transamination of ornithine) for detoxification of ammonia to glutamine; and 2. excretion of the glutamine thus formed as phenylacetylglutamine (in combination with phenylacetate) in the urine. This would have the advantage that the ammonia trapped as glutamine will not be available for later return to the circulation, resulting in net removal and reduction in ammonia concentration. This may lead to reduced circulating glutamine and resulting reduce gut production of ammonia, resulting in attenuation in systemic hyperammonemia.
In two recent experimental studies we proved the concept of this hypothesis. l ornithine phenylacetate adminstration attenuated the occurrence of hyperammonemia and elevated intracranial pressure in a devascularised pig model of acute liver failure (Ytrebø et al. 2007) and was able to reduce hyperammonemia and improved brain osmolytes and reduces brain water in a bile duct ligated rat model of cirrhosis (Wright et al. 2007). Furthermore, in liver disease patients, the safety of l-ornithine has been previously demonstrated. Also, phenylacetate/phenylbutyrate is considered safe and beneficial as an intervention to reduce hyperammonemia in urea cycle disorders. Therefore, the potential to test l-ornithine-phenylacetate in patients may only be a short series of confirmatory experiments away.
References
Bessman SP, Bradley JE (1955) Uptake of ammonia by muscle. Its implications in ammoniagenic coma. N Engl J Med 253:1143–1147
Clemmesen JO, Kondrup J, Ott P (2000) Splanchnic and leg exchange of amino acids and ammonia in acute liver failure. Gastroenterology 118:1131–9
Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P (1999) Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 29:648–53
Cooper AJL, Plum F (1987) Biochemistry and physiology of brain ammonia. Physiol. Rev 67:440–519
Cooper AJL, McDonald JM, Gelbard AS, Gledhill RF, Duffy TE (1979) The metabolic fate of 13N-labeled ammonia in rat brain. J Biol Chem 254:4982–4992
Dejong CHC, Kampman MT, Deutz NEP, Soeters PB (1992) Altered glutamine metabolism in rat portal drained viscera during hyperammonemia. Gastroenterology 102:936–948
Dejong CHC, Deutz NEP, Soeters PB (1993a) Renal ammonia and glutamine metabolism during liver insufficiency-induced hyperammonemia in the rat. J Clin Invest 92:2834–2840
Dejong CHC, Deutz NEP, Soeters PB (1993b) Metabolic adaptation of the kidney to hyperammonemia during chronic liver insufficiency in the rat. Hepatology 18:890–902
Ganda OP, Ruderman NB (1976) Muscle nitrogen metabolism in chronic hepatic insufficiency. Metabolism 25:427–435
Halperin ML, Kamel KS, Ethier JH, Stinebaugh BJ, Jungas RL (1992) Biochemistry and physiology of ammonium excretion, 2nd ed. Raven Press Ltd, New York
Haussinger D (1986) Regulation of hepatic ammonia metabolism: the intercellular glutamine cycle. Adv Enzyme Regul 25:159–80
Haussinger D, Kaiser S, Stehle T, Gerok W (1988) Structural and functional organization of hepatic ammonia metabolism: pathophysiological consequences. In: (ed) Advances in ammonia metabolism and hepatic encephalopathy. Excerpta Medica, Amsterdam, the Netherlands, pp 26–36
Jalan R, Wright G, Davies NA, Hodges SJ (2007) L-Ornithine phenylacetate (OP): a novel treatment for hyperammonemia and hepatic encephalopathy. Med Hypotheses 69(5):1064–9
Jalan R, Olde Damink SWM, Deutz NEP, Lee A, Hayes PC (1999) Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 354:1164–8
Kaiser S, Gerok W, Haussinger D (1988) Ammonia and glutamine metabolism in human liver slices: new aspects on the pathogenesis of hyperammonaemia in chronic liver disease. Eur J Clin Invest 18:535–42
Kircheis G, Nilius R, Held C, Berndt H, Buchner M Gortelmeyer R et al (1997) Therapeutic efficacy of l-ornithine-l-aspartate infusions in patients with cirrhosis and hepatic encephalopathy: results of a placebo-controlled, double-blind study. Hepatology 25:1351–1360
Lacey JM, Wilmore DW (1990) Is glutamine a conditionally essential amino acid? Nutr Rev 48:297–309
Lockwood AH, McDonald JM, Reiman RE, Gelbard AS, Laughlin JS, Duffy TE et al (1979) The dynamics of ammonia metabolism in man. Effects of liver disease and hyperammonemia. J Clin Invest 63:449–60
Lund PA (1970) radiochemical assay for glutamine synthetase, and activity of the enzyme in rat tissue. Biochem J 118:35–39
Meijer AJ, Lamers WH, Chamuleau RAFM (1990) Nitrogen metabolism and ornithine cycle function. Physiol Rev 70:701–748
Nelson D, Rumsey WL, Erecinska M (1992) Glutamine catabolism by heart muscle. Biochem J 282:559–564
Olde Damink SWM, Dejong CHC, Deutz NEP, van Berlo CLH, Soeters PB (1999) Upper gastrointestinal bleeding: an ammoniagenic and catabolic event due to the total absence of isoleucine in the haemoglobin molecule. Med. Hypotheses 52:515–519
Olde Damink SWM, Deutz NEP, Dejong CHC, Soeters PB, Jalan R (2002a) Interorgan ammonia metabolism in liver failure. Neurochem Int 41(2–3):177–88
Olde Damink SW, Jalan R, Redhead DN, Hayes PC, Deutz NE, Soeters PB (2002b) Interorgan ammonia and amino acid metabolism in metabolically stable patients with cirrhosis and a TIPSS. Hepatology 36(5):1163–71
Olde Damink SWM, Jalan R, Deutz NEP, Redhead DN, Dejong CHC, Hynd P, Jalan RA, Hayes PC, Soeters PB (2003) Hyperammonemia following a simulated or actual upper gastrointestinal bleed in patients with cirrhosis of the liver is predominantly caused by enhanced renal ammoniagenesis. Hepatology 37:1277–85
Owen EE, Johnson JH, Tyor MP (1961) The effect of induced hyperammonemia on renal ammonia metabolism. J Clin Invest 40:215–221
Owen OE, Reichle FA, Mozzoli MA, Kreulen T, Patel MS, Elfenbein IB et al (1981) Hepatic, gut, and renal substrate flux rates in patients with hepatic cirrhosis. J Clin Invest 68:240–52
Owen OE, Mozzoli MA, Reichle FA, Kreulen TH, Owen RS, Boden G et al (1985) Hepatic and renal metabolism before and after portasystemic shunts in patients with cirrhosis. J Clin Invest 76:1209–17
Plauth M, Roske AE, Romaniuk P, Roth E, Ziebig R, Lochs H (2000) Post-feeding hyperammonaemia in patients with transjugular intrahepatic portosystemic shunt and liver cirrhosis: role of small intestinal ammonia release and route of nutrient administration. Gut 46:849–55
Plumley DA, Souba WW, Hautamaki D, Martin TD, Flynn TC, Rout R et al (1990) Accelerated lung amino acid release in hyperdynamic septic surgical patients. Arch Surg 125:57–61
Smith I (1981) The treatment of inborn errors of the urea cycle. Nature 291:378–38
Souba WW, Klimberg VS, Plumley DA, Salloum RM, Flynn TC, Bland KI et al (1990) The role of glutamine in maintaining a healthy gut and supporting the metabolic response to injury and infection. J Surg Res 48(4):383–91
Staedt U, Leweling H, Gladisch R, Kortsik C, Hagmuller E, Holm E (1993) Effects of ornithine aspartate on plasma ammonia and plasma amino acids in patients with cirrhosis. A double-blind, randomized study using a four-fold crossover design. J Hepatol 19:424–430
Tyor MP, Owen EE, Berry JN, Flanagan JF (1960) The relative role of extremity, liver and kidney as ammonia receivers and donors in patients with liver disease. Gastroenterology 39:420–424
Walser EM, Harris VM, Harman JT, Park HM, Siddiqui AR (1996) Quantification of intrahepatic portosystemic shunting after placement of a transjugular intrahepatic portosystemic shunt. J Vasc Interv Radiol 7:263–7
Warter JM, Brandt C, Marescaux C, Rumbach L, Micheletti G, Chabrier G et al (1983a) The renal origin of sodium valproate-induced hyperammonemia in fasting humans. Neurology 33:1136–1140
Warter JM, Marescaux C, Brandt C, Rumbach L, Micheletti G, Chabrier G et al (1983b) Sodium valproate associated with phenobarbital: effects on ammonia metabolism in humans. Epilepsia 24:628–633
Weber FL, Veach GL (1979) The importance of the small intestine in gut ammonium production in the fasting dog. Gastroenterology 77:235–240
Webster LT, Gabuzda GJ (1958) Ammonium uptake by the extremities and brain in hepatic coma. J Clin Invest 37:414–424
Webster LT, Davidson CS, Gabuzda GJ (1958) Effect on portal blood ammonium of administering nitrogenous substances to patients with chronic hepatic disease. J Lab Clin Med 52:501–514
Welbourne TC (1988) Role of the lung in glutamine homeostasis. Contrib Nephrol 63:178–182
Welbourne TC, Phromphetcharat V, Givens G, Joshi S (1986) Regulation of interorgan glutamine flow in metabolic acidosis. Am J Physiol 250:E457–463
Welters CFM, Deutz NEP, Dejong CHC, Soeters PB (1999) Enhanced renal vein ammonia efflux after a protein meal in the pig. J Hepatol 31:489–496
Windmueller HG (1982) Glutamine utilization by the small intestine. Adv Enzym 53:201–237
Wright GA, Davies NA, Ytrebø LM, Stadlbauer V, Fuskevåg OM, Zwingmann C, Hodges S, Jalan R (2007) L-ornithine phenylacetate reduces arterial ammonia, improves brain osmolytes and reduces brain water in a bile duct ligated rat model of cirrhosis. Hepatology 46:604A (abstract)
Ytrebø LM, Kristiansen RG, Fuskevåg OM, Mæhre H, Revhaug A, Jalan R, Rose C (2007) L-ornithine phenylacetate attenuates increase in arterial ammonia and intracranial pressure in a devascularised pig model of acute liver failure: a novel ammonia-lowering strategy for hepatic encephalopathy. Hepatology 46:275A (abstract)
Ytrebø LM, Sen S, Rose C, Ten Have GA, Davies NA, Hodges S, Nedredal GI, Romero-Gomez M, Williams R, Revhaug A, Jalan R, Deutz NE (2006) Interorgan ammonia, glutamate, and glutamine trafficking in pigs with acute liverfailure. Am J Physiol Gastrointest Liver Physiol 291:G373–81
Zieve L (1987) Pathogenesis of hepatic encephalopathy. Metab Brain Dis 2:47–165
Acknowledgements
Steven W.M. Olde Damink was supported by a clinical fellowship from the Netherlands organization for health research and development (grant 907-06-177).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Olde Damink, S.W.M., Jalan, R. & Dejong, C.H.C. Interorgan ammonia trafficking in liver disease. Metab Brain Dis 24, 169–181 (2009). https://doi.org/10.1007/s11011-008-9122-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-008-9122-5