
Abstract
Impaired PI3K/Akt signaling (insulin resistance) and poor glycemic control (hyperglycemia) are the major risk factors involved in the progression of diabetic nephropathy (DN). This study was designed to identify factors influencing cell survival during DN. We found that high glucose exposure in renal proximal tubular cells (NRK52E) upregulated PHLPP1, an Akt phosphatase (Ser473), causing suppression in Akt and IGF1β phosphorylation leading to inhibition in insulin signaling pathway. Results demonstrate that sustained activation of PHLPP1 promoted nuclear retention of FoxO1 by preventing its ubiquitination via Mdm2, an Akt/ Nrf2-dependent E3 ligase. Thus, enhanced FoxO1 nuclear stability caused aberration in renal gluconeogenesis and activated apoptotic cascade. Conversely, gene silencing of PHLPP1-enhanced Nrf2 expression and attenuated FoxO1 regulated apoptosis compared to hyperglycemic cells. Mechanistic aspects of PHLPP1–Nrf2/FoxO1 signaling were further validated in STZ-nicotinamide-induced type 2 diabetic Wistar rats. Importantly, we observed via immunoblotting and dual immunocytochemical studies that treatment of Morin (2′,3,4′,5,7-Pentahydroxyflavone) during diabetes significantly augmented FoxO1 nuclear exclusion, resulting in its ubiquitination via Akt–Nrf2/Mdm2 pathway. Furthermore, lowering of PHLPP1 expression by Morin also prevented FoxO1/Mst1-mediated apoptotic signaling in vitro and in vivo. Morin treatment under the experimental conditions, effectively decreased blood glucose levels, ameliorated insulin resistance, alleviated oxidative stress and attenuated renal apoptosis in diabetic rats comparable to metformin thereby exhibiting tremendous potential against renal complications of diabetes. These novel results further acclaim that inhibition of PHLPP1/FoxO1–Mdm2 axis is critical in the pathogenesis of diabetic nephropathy.
Similar content being viewed by others


Introduction
Perturbation in the cell survival mechanisms has been found as the underlying cause in the pathophysiology of diabetic nephropathy [1]. Even with an increased understanding of pathways involved in apoptotic cell loss in diabetes, molecular cross talks involved in activating the apoptotic cascade have been scarcely studied. Amongst the other forms of chronic kidney disease, diabetic kidney disease (DKD) is intricately associated with insulin signaling aberration, tubular degeneration, hyperglycemia-mediated intra-renal ROS generation and renal cell death [2]. There is increasing evidence of higher mortality rate from the end-stage renal disease than that for other associated diabetic comorbidities [3, 4], illustrating an urgency to combat the complication. Kidneys play a major role in checking systemic glucose metabolism through regulation of glucose reabsorption, gluconeogenesis and glycolysis [5]. Emerging evidence show that factors involved in insulin signaling including, PI3K/Akt kinases, insulin receptor substrate 2 (IRS2), and Forkhead box protein’s (FoxO’s) are chief molecular players in the pathogenesis of DN [6]. FoxO1, FoxO3, FoxO4 and FoxO6 are four different members of the FoxO family which are regulated through several post-translational modifications. Phosphorylation, acetylation and ubiquitination are three major modifications that affect FoxO’s nuclear–cytosolic shuttling and stability. Multiple kinases including Akt [7, 8], CK1 (casein kinase 1), Inhibitory kappa B kinase (IĸB kinase), p38, AMPK (5' AMP-activated protein kinase), JNK (c-Jun N-terminal kinases), SGK (serum- and glucocorticoid-induced kinase), Mst1 (mammalian Ste20-like kinase 1) and CDK2 (cyclin-dependent kinase 2) [9] catalyze their phosphorylation thereby influences nuclear–cytoplasmic shuttling of FoxO proteins. Insulin signaling induces FoxO1 and FoxO3 phosphorylation via PI3K/Akt-dependent pathway, leading to their nuclear exclusion, ubiquitination and subsequent proteasomal degradation [10, 11].
In mammalian cells, FoxOs are degraded proteasomally, although the specific ubiquitin E3 ligases for FoxO proteins remain to be completely defined. Phosphorylation by Akt, IκB kinase (IKK) and ERK renders FoxOs susceptible to ubiquitination by E3 ligases including SKP2 (S-phase kinase-associated protein 2) and Mdm2 (Mouse double minute 2 homolog) [12] suggesting crucial role played by E3 ligases in regulating FoxOs activity. Mdm2 is an oncogene and is transcriptionally regulated by Nrf2 [13], which further integrates into p53 stabilization [14] and strongly influences cell survival. Although, the direct intervention of Nrf2 in regulating FoxO1 stability has not been addressed so far. We have recently reported that activation of PHLPP1 (Pleckstrin homology domain and leucine rich repeat protein phosphatase 1) suppresses Akt activation via its dephosphorylation at Ser473 residue which consequently limits Nrf2 responses and insulin signaling pathway during diabetic nephropathy [15, 16]. In addition, defects in Akt2 activation, have been reported in insulin-resistant type 2 diabetic and obese participants due to overexpression of PHLPP1 [17] and loss of PHLPP1 is known to enhance promoter acetylation of the receptor tyrosine kinase (RTK) in cancers [18]. These pieces of evidence provide a unique picture of PHLPP1 in the pathogenesis of type 2 diabetes and allow us to speculate if PHLPP1 could regulate ubiquitination of FoxO1 via Nrf2-mediated suppression of Mdm2 during DN.
In addition, there are reports suggesting that high glucose and diabetic conditions decrease FoxO1-dependent G6PD activity in endothelial cells, kidney, liver, and red blood cells which triggers oxidative damage leading to cellular dysfunction and cell death [19, 20]. Moreover, hyperglycemic state impairs the mitochondrial anti-oxidative enzymes causing mitochondrial dysfunction, mitophagy and apoptosis [21]. Uncontrolled mitochondrial ROS (mtROS) induces adverse effects in DN, causing hypertrophy, mesangial matrix expansion, podocyte apoptosis, and inflammation [22]. Plant derived phytochemicals are gaining interest in disease management due to their selective and high therapeutic capacity with minimal or no side effects. Therefore, we evaluated the effect of a bioflavonoid, Morin (2′,3,4′,5,7-pentahydroxyflavone) for its potential to modulate FoxO1 stability during diabetes. Morin has been extensively studied for its anti-oxidative [23,24,25,26], anti-inflammatory, anti-hyperglycemic [27] and insulin mimetic capacities [28]. Previous studies from our lab have demonstrated Morin as a strong inducer of Nrf2 [23, 24]. Morin also inhibits PHLPP protein by directly interacting with its PP2C domain [23]. It was thus considered more significant for this study, where we aim to identify its hypoglycemic effect through modulation in the PHLPP1–Nrf2/FoxO1 axis. Metformin is an oral hypoglycemic agent, widely used for the treatment of type 2 diabetic patients. Interestingly, it has been reported as a strong modulator of FoxO1 signaling [29,30,31]. In view of this, the effects of Morin alone or combined with metformin on diabetic nephropathy were investigated. Also, the combination of Morin and Metformin was evaluated herein for its efficacy to prevent Metformin-induced side effects that incur amongst diabetics. Importantly, FoxO1 inhibition has been demonstrated to have beneficial effects on glucose homeostasis and insulin sensitivity in insulin resistant animal models [32, 33]. In this study, we aimed to unravel the mechanism that stabilizes nuclear FoxO1 nuclear causing aberrant glucose influx and renal apoptosis during diabetes. Thus, exploration of a novel role of PHLPP1 in activating FoxO1 during hyperglycemia may provide a better understanding of glucose metabolism at the molecular level and shall allow the development of therapeutic strategies.
Materials and Methods
Antibodies including FoxO1-C29H4 (2880), Phospho-FoxO1 (Ser256) (9461), phospho-Akt Thr308 (2965), Pan Akt (4691P), phospho-MDM2 Ser 166 (3521), Histone (h3) (4499 s), Mst1(3682), phospho-Mst1 Thr183 (3681), Bim (C34C5) Rabbit mAb, 2933, anti-rabbit Alexa-fluor 555 conjugate (4413), anti-goat FITC conjugated secondary antibody (F7367), and anti-rabbit FITC conjugated secondary antibody (F7367) were purchased from Cell Signaling Technology (Danvers, MA, USA). Chemicals and antibodies like Nrf2 (sc-13032), β-actin (sc-81178), Ubiquitin (sc-166553), HO1 (sc-10789), NQO1 (sc-16463), caspase-3 (sc-7148), Bax (sc-493), Bcl2 (sc-492-G), p-Akt1/2/3 Ser 473 (sc-101629) and Protein A/G PLUS Sepharose beads (sc-2003) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Heat-inactivated fetal bovine serum was purchased from Gibco Life Technologies Ltd. (Grand Island, NY, USA. 100X antimycotic and antibiotic solution, OPTI-MEM reduced serum medium, Lipofectamine RNAi MAX Transfection Reagent and Hoechst 33,258 was procured from Invitrogen (Carlsbad, CA. USA). All other reagents or chemicals including streptozotocin, nicotinamide, PHLPP1 antibody (SAB1300919) were procured from Sigma (St Louis, MO, USA) unless otherwise mentioned.
Animal treatments
All experiments on rats were conducted in accordance with the international guidelines and protocols approved by the Institutional Animal Ethics Committee (IAEC) of CSIR-IITR with approval no. IITR/IAEC/01/16–82/16. Total 30 male Wistar rats (4–5 weeks) were procured from IITR animal house and were kept in experimental rooms with controlled temperature (23 ± 2 °C), humidity (40–60%) and regular light/ dark cycles of 12 h each. Rats were maintained on a standard pellet diet (Altromin International, Germany) and water ad libitum.
Induction of type II diabetes and treatments
30 rats were randomly divided into five groups of six animals each (n = 6). Diabetes (hyperglycemia) was induced in rats by single intraperitoneal injection of streptozotocin (STZ; 60 mg/kg) in cold sodium citrate buffer (0.1 M, pH 4.5) as described by Masiello et al., (1998) [34]. Rats were injected with nicotinamide (120 mg/kg) in normal saline, to prevent excessive pancreatic damage and establish hyperglycemic conditions. Nicotinamide was injected 15 min prior to STZ, as per the protocol described earlier by our group [35]. Streptozotocin is an antibiotic derived from Streptomyces achromogenes and a structural glucosamine derivative of nitrosourea, which shows selective toxicity in pancreatic β-cells and induces diabetes. It is readily taken up by β-cells through glucose transporter GLUT-2 which produces excessive free radicals causing DNA damage, followed by activation of nuclear enzyme poly(ADP-ribose) synthetase, an enzyme involved in DNA repair. Elevated activity of the enzyme depletes its substrate nicotinamide adenine dinucleotide (NAD), resulting in reduced insulin synthesis in β-cells and cell death. This streptozotocin-induced cytotoxicity is alleviated by nicotinamide, a component of NAD, which inhibits poly (ADP-ribose) synthetase activity and prevents NAD depletion in pancreatic β-cells [36, 37]. Therefore, the streptozotocin–nicotinamide model exhibits moderate hyperglycemia associated with loss of postprandial early phase insulin secretion and nearly 50% decrease in pancreatic insulin content which makes it the most suitable model for understanding hyperglycemic mechanisms.
Rats with blood glucose levels higher than 250 mg/dL after 3 days of STZ injection were considered diabetic and selected for further experimentation. No mortality was observed throughout the experimental period in all the groups.
Group I Control rats received normal saline.
Group II Rats injected with STZ/nicotinamide to induce diabetes.
Group III Diabetic rats treated with Morin (30 mg/Kg bwt./day) by oral gavage.
Group IV Diabetic rats treated with metformin (50 mg/Kg bwt./day) intra-peritoneally.
Group V Diabetic rats treated with metformin (50 mg/Kg bwt./day) + Morin (30 mg/Kg bwt./day).
Body weight of rats was monitored regularly throughout the experimental period (30 days). After completion of treatment, animals were sacrificed by euthanasia using animal ethics guidelines. Blood was drawn through cardiac puncture and collected in glass tube vials for efficient serum separation and centrifuged at 2000 rpm for 15 min to obtain clear serum. Subsequently, the serum was stored at –80ºC for biochemical studies. Immediately after blood collection, kidneys were harvested, rinsed in saline and weighed on an electronic balance (Uni Bloc, Shimadzu, Japan). A small portion weighing ~ 100 mg from each kidney was placed in the Trizol solution for RNA extraction. The remaining tissue was stored at –80 °C for further experimentations. Serum parameters including creatinine and urea were assessed on RANDOX autoanalyzer (Randox Laboratories Ltd., UK).
Metabolic study (OGTT)
To assess the metabolic function in rats, oral glucose tolerance test (OGTT) was performed in the same set of diabetic rats thrice after diabetes induction by giving 2 g/Kg bwt. of glucose to overnight fasted rats as described earlier [38]. Blood samples were collected from the rat tail vein at 30 min intervals for 2 h to assess blood glucose concentration using glucometer (Accusure, Tai Doc Technology, Taiwan). Data are represented as blood glucose concentration (mg/dL) at different time points (0–120 min).
Insulin ELISA assay
Serum insulin levels of treated and control rats were determined using insulin ELISA assay kit (Purgene, Geneix Biotech Asia Pvt. Ltd.). Insulin estimation was done according to the manufacturer’s protocol and the absorbance was read at 450 nm on Spectramax plus 384 microplate based UV/visible spectrophotometer (Molecular devices, CA, USA). Insulin levels are represented in ng/mL.
Oxidative stress assessment
Total Thiols
Total thiol levels were estimated by the method of Himmelfarb et al., 2000 [39]. To estimate the protein sulfhydryl content, 20 µl clear renal homogenate (500 µg protein) was reacted in a cuvette with 10 mM DTNB (in methanol), 0.1 M Tris buffer (pH 8.2) containing 10 mM EDTA. The absorbance was recorded at 412 nm after 10 min incubation on SpectraMax Plus384 Microplate Reader (Molecular Devices, CA, USA). The readings were normalized by absorbance of each sample blank, where DTNB (5,5'-dithio-bis-(2-nitrobenzoic acid) was excluded from the reaction. The results are expressed as total thiol content/mg protein.
Lipid peroxidation
Lipid peroxidation was estimated as described by the method of Niknahad et al., 2003 [40]. Briefly, to 50μL (~ 5 mg) of fresh renal tissue homogenates, added 50 μL of distilled water, 25μL of 40%TCA and 100μL of 1.3% thiobarbituric acid (TBA). Subsequently, the samples were incubated in a water bath at 100 °C for 20 min and thereafter the reaction was stopped immediately by placing the samples on ice. Samples were then centrifuged at 2600 rpm for 10 min. The absorbance of the supernatant was read at 530 nm and 630 nm on SpectraMax Plus384 Microplate Reader (Molecular Devices, CA, USA). Thiobarbituric acid reactive substance (TBARS) was calculated using 1,1’,3,3’ tetramethoxy propane as standard. The results are represented as nM TBARS/mg protein.
Estimation of enzyme activities
Catalase activity
Catalase enzyme converts hydrogen peroxide to water and oxygen thereby protects the cell from oxidative damage by reactive oxygen species. Catalase activity was assayed in renal tissue lysate by the method of Sinha et al., [41]. To freshly prepared 10μL of renal lysate, added 100μL of 0.1 M potassium phosphate buffer (pH 7.0) followed by 40μL of 2 M H2O2, whereas respective blanks lacked hydrogen peroxide. Immediately after 10 s, 150 μL stopping reagent [5% Potassium dichromate and glacial acetic acid; (1:3)] was added and the absorbance was read at 530 nm on Spectramax plus 384 microplates-based UV/visible spectrophotometer (Molecular Devices, CA, USA). The values were subtracted from respective blanks, where stopping reagent was added prior to H2O2 and the activity is represented as µM H2O2 consumed/min/mg protein.
Thioredoxin reductase activity
Thioredoxin reductase enzyme catalyzes the reduction of thioredoxin and is a central component in the thioredoxin system. Electrons are taken from NADPH via thioredoxin reductase and are transferred to the active site of thioredoxin, which goes on to reduce protein disulfides or other substrates. The enzyme activity was estimated following the method of Ellman, 1959 [42]. Thioredoxin reductase catalyzes the reduction of oxidized thioredoxin while converting DTNB to TNB (5 thio-2-nitrobenzoic acid) in the reaction. Briefly, the method involves the addition of 500μL of phosphate buffer [500 mM (pH 7.5)] with NADPH (1 mM) to 10 µl of renal lysate (~ 500 µg). The reaction initiated as soon as DTNB (8 mM) was added in the mixture. The kinetic absorbance was taken at 412 nm at 30 s intervals for 3 min. Sample absorbance was subtracted by reagent blank and the sample blank which lacked DTNB. The absorbance was taken on Ultrospec 3100pro UV/visible spectrophotometer (Amersham Biosciences, Sweden). Enzyme activity was calculated as µM TNB formed/min/mg protein (Extinction coefficient of TNB = 13,600 M−1 cm−1) and represented as fold change compared to control.
Glutathione Reductase (GR) activity
Glutathione reductase activity was assessed according to the method by Mannervik, 1999 [43]. GR activity is measured as the amount of NADPH consumed in reducing GSSG to GSH. The assay was performed in a cuvette containing fresh renal homogenate with 500 µg protein (10 μl), 1 mM GSSG, 0.2 mM NADPH and 0.1 M Tris–HCl buffer (pH 7.5). The absorbance was read spectrophotometrically on Ultrospec 3100pro UV/visible spectrophotometer at 340 nm for 3 min at 30 s intervals. Results are expressed as fold change of control using extinction coefficient for NADP+ = 6.22 mM−1 cm−1.
Cell culture and treatments
Normal rat kidney cell line NRK52E (ATCC, Manassas, VA, USA) was cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 2 mM glutamine (Gibco), 3.7 g/L sodium bicarbonate, 10% FBS, 1% antibiotic–antimycotic solution under standard conditions of 37 °C in humidified atmosphere and 5% CO2 in an incubator (Thermo-Forma, model no. 371).
Hyperglycemia was induced in NRK52E cells by subjecting them to 30 mM glucose for 48 h. To assess the protective effect of Morin, hyperglycemic cells were treated with 30 µM Morin and metformin (50 μM) was used as standard drug control. Doses for Morin and metformin were selected on the basis of MTT cytotoxicity assay (Supplementary Fig. S1). To delineate the role of Akt impairment in Nrf2/FoxO1 signaling, we used specific PI3K/Akt inhibitor (LY29002, Sigma, MO, USA) at a concentration of 30 µM (24 h), based on MTT assay.
ROS estimation
DCFHDA assay
Total ROS formation was estimated using fluorescent probe DCFH-DA (2′,7-dichlorofluorescein di-acetate) in black coated 96 well plates (1X104 cells/well) according to the method by Zhang et al., 2009 [44]. 10 μM of DCFHDA was added in each well after completion of the treatment period and was allowed to incubate for 30 min at 37ºC. Upon entering the cell, the diacetate bond of the fluoroprobe is cleaved by intracellular esterases leaving DCFH (Dichloro di hydrofluorescin) which is oxidized to DCF (dichlorofluorescein) by the oxidants and the fluorescence is taken as a marker of ROS production in the cell. The fluorescence intensity was measured at wavelengths with Ex/Em of 485/530 nm on a Varioskan fluorescent microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Estimation of superoxide radical generation
Dihydroethidium (DHE) assay was performed to specifically monitor intracellular superoxide radical generation as described by Zhao et al., [45]. Treatment was performed in black coated 96 well plate with a seeding density of 1X104 cells/well. DHE upon reaction with superoxide anions forms a red fluorescent product (ethidium) which intercalates with DNA. DHE is perhaps the most specific dye as it detects essentially superoxide radicals and is retained well by cells. After indicated treatment schedule, cells were incubated with 10 μM DHE for 30 min at 37 °C and the fluorescence of living cells blue (Ex/Em 370/420 nm) and chromatin of living cells red (Ex/Em 535/610 nm) was taken on Varioskan fluorescent microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). Results are expressed as Ethidium/DHE ratio.
Estimation of cellular GSH levels (CMFDA)
5-chloromethyl fluorescein diacetate, CMFDA (Molecular Probes, Eugene, Oregon, USA) is a CellTracker green dye used to analyze the GSH content in the cells with a specificity of 95% [46]. The CMFDA dye contains a chloromethyl group which passes freely through the cell membrane and reacts with thiols, in a glutathione S-transferase-mediated reaction. Briefly, cells were seeded in 96 black well plate and after completion of treatment schedule, cells were incubated with 5 µM of CMFDA for 30 min at 37 °C. The fluorescent adduct was detected at (Ex/Em) 492/517 nm on a Varioskan fluorescent microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
RNA preparation and qRT-PCR
FoxO1-dependent gluconeogenic gene expression was studied using real-time PCR analysis for which total RNA was extracted from treated renal cells samples (NRK52E) using trizol (Thermo Scientific, MA, USA) and RNA concentrations were determined spectrophotometrically (A260/280). Real-time PCR analysis was performed on Quant studio 6 flex, Applied Biosystems, (Thermo Fisher Scientific Inc., MA, USA) using 2X SYBR Green Supermix (Thermo Fisher Scientific Inc., MA, USA). The sequences of rat primers used for different genes are:
G6Pase F 5′GGACCTCCTGTGGACTTTGG 3′R 5′AAACGGAATGGGAGCGACTT3′
G6K F 5′CCTGGGAGGAACCAACTTCA 3′R 5′TCTTGTGCTTCATCATCTCGGC3′
PEPCK F 5′TGCCCAAGATCTTCCACGTC 3′R 5′TCAAGTTCAGGGCGTCTTCC3′
β-Actin F 5′ TGAGAGGGAAATCGTGCGT 3′ R 5′ TCATGGATGCCACAGGATTCC3′
Gene expression was analyzed using the comparative cycle time (2−△△Ct) method and the target gene expression was normalized to β-actin (endogenous reference).
Gene knockdown of PHLPP1 by si RNA
Gene silencing for PHLPP1 was performed using Silencer Select predesigned siRNA (ID: s133147) procured from Ambion (Austin, TX, USA). Negative siRNA (Am4611, Austin, TX, USA) was taken as the negative control. Transfection was performed in agreement with the manufacturer's instructions using Lipofectamine RNAiMAX transfection reagent. Briefly, after 24 h of plating, cells were transfected with 50 nM of the silencer in Opti-MEM reduced serum medium (Invitrogen, Life Technologies Corp., USA). After 12 h incubation, the transfection medium was replaced with serum supplemented DMEM and cells were further cultured for an additional 48 h under hyperglycemic conditions. Thereafter, western blotting analysis was performed to confirm knockdown of PHLPP1 compared with negative control siRNA (scrambled siRNA).
Immunoblotting
Renal tissue/cell proteins were isolated by the method as described earlier in Mathur et al., 2018 [16]. Initially, tissue/cells were washed twice with 1X PBS followed by lysis in NP-40 buffer using 1X protease and phosphatase inhibitor cocktails. The lysate was centrifuged at 17,000 × g at 4 °C for 15 min. The supernatant containing proteins was collected and the concentration of protein in the lysate was estimated by BCA method (Bicinchoninic acid assay).
The gel for protein samples was run at 120 V for nearly 3–4 h and subsequently, the gel was transferred on PVDF membrane (Hydrophobic Immobilon-P, Millipore, MA, USA) at 100 V for 3 h. The blots were incubated in 1X blocking buffer (Sigma, MO, USA) for 1 h. The blots were washed with 1X TBST and incubated with 1° antibodies (1:1000) for 4 h and with 2° antibody (1:2000) for 3 h on the shaker. Western blots were developed on Amersham Imager 600 detection system (GE Healthcare, UK) using Immobilon Western chemiluminescent horseradish peroxidase substrate kit (Millipore, MA, USA). The protein levels were quantified and analyzed by ImageJ 1.44p software (National Institute of Health, Bethesda, MD, USA). The procedure was performed for various signaling proteins including PHLPP1, FoxO1, p-FoxO1 (Ser256), FoxO1, p-Akt (Thr308), p-Akt (Ser473), Pan Akt, p-MDM2 (Ser 166), Mst1(3682), p-Mst1(Thr183), Bax, Bim, Bcl2, Caspase3, p-IGF1β (Tyr1135/1136), Nrf2, NQO1, HO-1, Histone (h3) and β-actin.
Co-immunoprecipitation study
Renal tissue (~ 100 mg) was homogenized in non-denaturing NP-40 buffer (20 mM HEPES, pH 7.4, 1% NP-40, and 1 mM DTT). For the experiment, the lysate (~ 500 µg) was primarily incubated for 12 h with the p-Mdm2/FoxO1 antibody on rocker after which, Protein A/G PLUS sepharose beads were added and again incubated for 6 h at 4 °C on the rocker. The beads were rinsed thrice with chilled NP-40 buffer and boiled in 2X SDS loading dye. Samples were then run on SDS-PAGE for immunoblotting as described above.
Propidium iodide (PI) uptake assay
Propidium Iodide is a red fluorescent membrane dye used to assess cell death depending upon membrane integrity as PI internalizes in cells having damaged membranes. Assessment of cell death was performed by seeding 10,000 cells per well in 96 well black coated plates and incubated overnight, followed by high glucose/ Morin and metformin treatment. After 48 h of treatment, cells were incubated with propidium iodide (1 mg/mL) at 37ºC for 30 min. Fluorescence was measured on Varioskan fluorescent microplate reader at Ex/Em 535/610 nm (Thermo Fisher Scientific, Waltham, MA, USA).
Glucose uptake
2-NBDG is a fluorescent deoxyglucose analog that can be taken up by cells through glucose transporters. Cells were treated with 100 µg/mL of 2-NBDG for 30 min after completion of treatment and the uptake was calculated by measuring fluorescence at wavelengths of Ex/Em 485/528 nm on Varioskan fluorescent microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Statistical analysis
All computational calculations of quantitative data were performed using the Microsoft Excel program. Each experiment was repeated at least three times. Values are mean ± SE of observations. Statistical comparisons between means of different groups were conducted by one-way Analysis of Variance (ANOVA). Differences were considered statistically significant when p < 0.05.
Results
Hyperglycemia induces PHLPP1-mediated dephosphorylation of Akt at Ser-473 and prevents FoxO1 phosphorylation
Inactivation of the PKB/Akt pathway is a key feature observed during impaired insulin signaling pathway [7, 47, 48]. Initial experiments were undertaken to determine the levels of PHLPP1, a phosphatase for Akt (Ser 473), in renal tissues of treated Wistar rats. Figure 1a demonstrates significant up-regulation of PHLPP1 (2.26-fold; p < 0.05) in diabetic rats resulting in decreased Akt phosphorylation at Ser473 to 0.62-fold (p < 0.05) compared to control rats (Fig. 1a, b). This was further accompanied by lowered phosphorylation of insulin growth factor (IGF-1β, Tyr 1135/1136) during diabetes directing towards obstructed insulin signaling pathway. The decrease in Akt activation prevented FoxO1 phosphorylation during diabetes suggestive of suppressed FoxO1 ubiquitination and degradation. Treatment with Morin (30 mg/Kg bwt./day) in diabetic rats significantly suppressed PHLPP1 levels by 0.6-fold and enhanced Akt phosphorylation (1.17-fold; p < 0.05) compared to diabetic rats (Fig. 1a, b). Anti-diabetic effects of Morin were compared with Metformin as it is known to modulate FoxO1 activity. Results demonstrate that activation of Akt by Morin significantly upregulated FoxO1 phosphorylation at Ser 256 (1.25-fold; p < 0.05) compared to diabetic and metformin (0.61; p < 0.05) treated rats (Fig. 1a, b). Co-treatment of Morin and Metformin was also administered to a set of diabetic rats to assess if Morin could prevent metformin associated reno-toxicity in vivo. It was observed that co-treatment (Mo + Met) in diabetic rats significantly up-regulated insulin signaling compared to diabetic group as FoxO1 phosphorylation was enhanced up to 1.26-fold; p < 0.05 along with p-IGF1β levels (1.57-fold; p < 0.05), whereas metformin treatment could activate p-IGF1β up to 0.75-fold (p < 0.05) as compared to Morin treated rats suggesting better reno-protective efficacy of Morin via modulation in PHLPP1/FoxO1 axis (Fig. 1a, b). To validate if PHLPP1 induction occurs specifically under hyperglycemic milieu, we exposed renal tubular epithelial cells (NRK52E) to high glucose (30 mM) for 48 h (Fig. 1c, d). Immunoblotting data revealed that hyperglycemia significantly up surged PHLPP1 levels compared to control (2.0-fold; p < 0.05) which subsequently rendered activation of FoxO1 via Akt inhibition (Fig. 1c, d). Similar to the results observed in vivo, Morin averted PHLPP1-mediated FoxO1 activation, resulting in enhanced IGF-1β phosphorylation (insulin action). Overall, the data imply that hyperglycemic conditions provoke elevation in phosphatase action of PHLPP1 which averts Akt-induced FoxO1 phosphorylation leading to aberrant insulin signaling pathway (Fig. 1c, d).

Hyperglycemia upregulated PHLPP1 signaling in renal cells. a Immunoblots of PHLPP1, p-Akt, FoxO1 and IGF1β in diabetic, Morin and/or metformin treated rat renal tissue lysate (in vivo). b Graph represents protein quantification of the immunoblots depicted in panel A. c Immunoblots for critical proteins including PHLPP1, Akt, p-FoxO1 and p-IGF1β in high glucose (30 mM), Morin and metformin treated NRK52E cell lysates (in vitro). β-actin was used as a loading control for the immunoblots. d Bar graph denotes densitometry of the western blots shown in panel C. Data are presented as mean ± SE *p < 0.05 vs control, #p < 0.05 vs hyperglycemia/diabetic
PHLPP1 regulates FoxO1 activation by suppressing Nrf2/Mdm2 expression
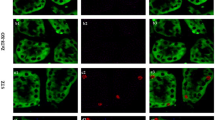
Insulin signaling normally limits FoxO1 activation via Akt-mediated phosphorylation on its Ser256 residue which promotes nuclear exclusion of FoxO1 and consequently stimulates cell survival [15, 49]. Consistent with the literature, we observed enhanced nuclear retention of FoxO1 in hyperglycemic NRK52E cells, while gene silencing of PHLPP1 significantly attenuated the nuclear stability of FoxO1 (Fig. 2a) implicating that PHLPP1 regulates FoxO1 translocation. Immunoblotting studies further reveal that p-FoxO1 levels were enhanced significantly to 1.45-fold (p < 0.05) in PHLPP1 silenced samples compared to the hyperglycemic group (Fig. 2b). This was accompanied by up-regulation in Akt phosphorylation (Ser473) confirming the role of PHLPP1/Akt axis in the regulation of FoxO1 (Fig. 2b). Interestingly, we observed lowered expression of the p-Mdm2 protein, an E3 ligase of FoxO1 in the hyperglycemia treated cells which implies that insufficient FoxO1 ubiquitination propels its nuclear retention under high glucose conditions (Fig. 2b). Reciprocally, abrogation of PHLPP1 through selective gene knockdown accentuated Akt-dependent phosphorylation of Mdm2 as observed through immunocytochemistry and immunoblotting in vitro (Fig. 2b, c, d). Moreover, Mdm2 regulatory protein, Nrf2, was also found to be lowered in the hyperglycemic cells, whereas PHLPP1 silenced cells show significantly increased levels of Nrf2 (Fig. 2b).

Gene silencing of PHLPP1 promoted inactivation of FoxO1. a Immunofluorescent staining of FoxO1 (FITC) in NRK52E cells subjected to hyperglycemic stress (30 mM; 48 h). Nuclear accumulation of FoxO1 was reduced by Morin (30 μM) and metformin treatment (50 μM). PHLPP1 silenced cells showed significant decrease in FoxO1 nuclear translocation. Images were captured at 20X magnification, scale bar 50 μm. PHLPP1 gene silencing was performed using silencer RNA. b Immunoblots of PHLPP1 targeted proteins including Akt, FoxO1, Nrf2 and Mdm2 were assessed in the cell lysates obtained after silencing PHLPP1 gene in NRK52E cells. β-actin served as a loading control. c Graph depicts densitometry of western blots shown in panel B. d Immunocytochemistryl of p-Mdm2 (Alexa Fluor) protein in NRK52E cells treated with high glucose, Morin, Metformin and PHLPP1 silencer (si-RNA) for 48 h. Images were captured at 20X magnification, scale bar 50 μm. e Graph depicts mean fluorescence intensity of p-Mdm2. Data are presented as mean ± SE *p < 0.05 vs control, #p < 0.05 vs hyperglycemia
Consistent with the reports that mention FoxO1 modulatory capacity of metformin, we observed that metformin-enhanced nuclear exclusion of FoxO1 compared to hyperglycemic cells (Fig. 2a). However, Morin effectively prevented FoxO1 nuclear retention compared to metformin treated cells. Overall, the observations support the fact that PHLPP1 increases FoxO1 nuclear stability via Nrf2/Mdm2 pathway during hyperglycemia, while Morin precludes it by declining PHLPP1 expression.
PHLPP1 impedes nuclear stability of Nrf2 and lowers its transcriptional activity in response to glucose toxicity
To further affirm the role of Nrf2/Mdm2 axis in regulating FoxO1 expression, we ascertained the expression of Nrf2 and its responsive genes in renal samples. Western blotting studies show decreased Nrf2, NQO-1 and HO-1 protein levels in vitro and in vivo under hyperglycemic conditions, while the levels were enhanced significantly by Morin treatment compared to high glucose and metformin treated cells (Fig. 3a–d). Nrf2 acts by binding to ARE (antioxidant response element) region in the nucleus and thereby transcribe its responsive genes. Therefore, we evaluated nuclear and cytosolic protein levels of Nrf2 in the treated kidney samples to affirm its action (Fig. 3e). Results show that the nuclear translocation of Nrf2 was significantly reduced in the sub-cellular fractions of diabetic renal tissue lysate compared to control, while Morin treatment significantly induced Nrf2 nuclear retention (Fig. 3e). These results show that suppression in Nrf2 during high glucose condition restrains its transcriptional efficiency. Moreover, immunocytochemistry in NRK52E cells show enhanced nuclear retention of Nrf2 by Morin compared to hyperglycemic cells (Fig. 3f, g).

Hyperglycemia suppresses PHLPP1-mediated Nrf2 responses in the kidney. a Immunoblots of Nrf2 and its down-regulated genes assessed in the renal tissue lysates of diabetic, Morin and/or Metformin treated rats. b Graph denotes densitometry of the immunoblots shown in panel A. c Immunoblots of Nrf2 and its regulated proteins NQO1 and HO1 in the NRK52E cell lysates treated with high glucose (30 mM), Morin and metformin for 48 h. β-actin was used as a loading control. d Bar graph denotes protein densitometry of the immunoblots shown in panel C. e Nuclear translocation of Nrf2 was evaluated in the subcellular fractions of nuclear and cytosolic lysates obtained from diabetic, Morin/metformin treated rats. Histone (H3) was used as a nuclear protein loading control, while β-actin served as the cytosolic loading control. f Immunocytochemistry of Nrf2 in NRK52E cells treated with high glucose, Morin and /or metformin for 48 h. Images were captured at 20X, scale bar 50 µm. g Graph shows the relative fluorescence intensity of Nrf2 in the treated cells. h Effect of Akt inhibition on FoxO1 signaling was studied using PI3K/Akt inhibitor, LY29002 (30 μM) for 24 h in NRK52E cells. Western blots of FoxO1, p-FoxO1, p-Mdm2 and PHLPP1 regulated Mst1 protein in inhibitor treated cell lysates. i The bar graph denotes protein densitometry of the immunoblots shown in panel H. Data are presented as mean ± SE *p < 0.05 vs corresponding control, #p < 0.05 vs hyperglycemia/diabetic
To validate modulation of FoxO1 by Akt–Nrf2/Mdm2 axis during hyperglycemia, we inhibited Akt activity in NRK52E cells with LY29002 (chemical inhibitor of PI3K/AKT), where immunoblotting data indicated significantly lowered phosphorylation of FoxO1 and p-Mdm2 upon Akt inhibition similar to hyperglycemic cells (Fig. 3h, i). On the contrary, Morin treatment enhanced the phosphorylation of FoxO1 indicating that suppression of Akt by PHLPP1 prevents FoxO1 ubiquitination (Fig. 3h, i). Furthermore, these results allowed us to infer that Morin promotes Nrf2 induction thereby enhances Mdm2-mediated FoxO1 degradation.
PHLPP1 promotes FoxO1 nuclear translocation by restricting its ubiquitination via Nrf2/Mdm2 axis
To identify the mechanism for enhanced FoxO1 activation during hyperglycemia, we performed co-immunoprecipitation studies, where ubiquitination of FoxO1 was assessed in renal samples. Reduced FoxO1 ubiquitination was observed in the diabetic group compared to control rats (Fig. 4a). Furthermore, diabetic conditions averted Mdm2 interaction with FoxO1 which probably restrained ubiquitin tagging on FoxO1 for degradation (Fig. 4b). Results show that enhancement of Mdm2 expression via Nrf2 in Morin treated renal samples promoted FoxO1 degradation significantly (Fig. 4a, b). Metformin treatment showed weak association between Mdm2 and FoxO1 compared to diabetic tissues. These results suggest that suppressed degradation of FoxO1 during diabetes accentuated its nuclear accumulation. A similar phenomenon was also observed in NRK52E cells, where the dual immunocytochemistry study shows lowered FoxO1 and ubiquitin interaction in the hyperglycemic cells compared to control cells, suggesting lowered FoxO1 degradation. On the other hand, Morin enhanced the interaction of FoxO1 with ubiquitin, promoting cell survival signaling (Fig. 4c). Specifically, we also assessed p-Mdm2 and FoxO1 protein levels in the nuclear and cytosolic sub-cellular fractions of kidney tissue lysates (Fig. 4d, e). Data demonstrate that under diabetic conditions, expression of FoxO1 was enhanced significantly in the nuclear compartment, whereas p-Mdm2 levels were lowered in the nuclear fraction of diabetic rats, lowering the possibility of their interaction. Interestingly, Morin treatment prevented the nuclear stabilization of FoxO1 resulting in its enhanced translocation in the cytosolic fraction. Hence, it can be conceived that loss of Nrf2/Mdm2 function promotes activation of FoxO1 during hyperglycemia causing progression in the pathogenesis of diabetic nephropathy.

PHLPP1 prevents Nrf2/Mdm2-mediated FoxO1 ubiquitination during hyperglycemia. a Western blot of co-immunoprecipitated treated samples to assess the interaction of FoxO1 with ubiquitin in vivo. Whole tissue lysates were used for co-immunoprecipiation from each treated samples. b Mdm2 protein was co-immunoprecipitated with the treated in vivo tissue lysates and samples were probed with FoxO1 on the immunoblot. c Dual immunocytochemistry was performed on NRK52E cells to study FoxO1 ubiquitination in hyperglycemic and Morin treated cells (FoxO1-FITC; Ubiquitin-Alexa Fluor). The images were captured at 100X, scale bar 10 μm. d Expression of FoxO1 and p-Mdm2 proteins was assessed through immunoblotting in the nuclear fraction of treated tissue lysates. Histone (h3) served as a loading control. e Immunoblots of FoxO1 and p-Mdm2 proteins in the cytosolic fraction of treated tissue lysates. β-actin served as the loading control for the cytosolic proteins. Data are presented as mean ± SE *p < 0.05 vs corresponding control, #p < 0.05 vs hyperglycemia/diabetic
Activation of FoxO1 impairs gluco-metabolic activity in renal cells
So far, the results demonstrate activation of FoxO1 during hyperglycemia and this activation has been identified as a critical mechanism behind impaired glucose tolerance. FoxO1 is known to coordinate gluconeogenesis by regulating the transcription of gluconeogenic genes including PEPCK (Phosphoenolpyruvate carboxykinase), G6K (Glucose 6 kinase) and G6P (Glucose-6-phosphatase) upon its nuclear translocation. Herein, we investigated the expression of FoxO1-dependent gluconeogenic genes through real time PCR. In contrast to the expected gene expression, we observed lowered m-RNA expression of FoxO1-dependent glycolytic genes in hyperglycemic NRK52E cells, while Morin supplementation restored the aberrant decline in gene expression of G6K (3.4-fold), G6Pase (2.3-fold) and PEPCK to 2.7-fold; (p < 0.05) compared to hyperglycemic cells (Fig. 5a). The unusual decline in gluconeogenic genes could be due to dual regulation by both the reabsorbed glucose via the SGLTs and the insulin signaling in proximal tubular cells. It has been reported that renal gluconeogenesis is suppressed, not only by insulin signaling but also due to glucose reabsorption during fed states [35]. The above statement was validated when we observed enhanced glucose uptake in the hyperglycemic NRK52E cells, while Morin, metformin reduced the uptake significantly (Fig. 5b). The elevation in glucose uptake under high glucose treatment affected the gluconeogenic gene expression. Therefore, these results reflect towards anomalous metabolic actions during hyperglycemia contributing to nephrotoxicity.

FoxO1 regulates gluco-metabolic genes during hyperglycemia in renal cells. a Transcriptional expression (m-RNA) of FoxO1 and its regulated metabolic genes in vitro. Expression of Glucose 6 kinase (G6K), Glucose 6 phosphatase (G6Pase) and phosphoenolpyruvate kinase (PEPCK) was assessed in renal cells (NRK52E) treated with high glucose (30 mM), Morin and/or metformin. Fold change in mRNA levels was calculated using 2−△△Ct method between samples, and the amount of target was normalized to β-actin as an endogenous reference. b Graph indicates 2-NBDG fluorescence, indicative of cellular glucose uptake in NRK52E cells treated with high glucose (30 mM), Morin (30 µM) and/or metformin (50 µM). Data are presented as the mean ± S.E.. *p < 0.05 vs corresponding control, #p < 0.05 vs hyperglycemia
Morin prevented PHLPP1-induced hyperglycemic renal toxicity in rats
Adequate insulin signaling is essential for glucose metabolism in kidneys. We observed that diabetic rats had elevated insulin levels in their blood serum samples (1.56-fold; p < 0.05) compared to control rats confirming the establishment of type 2 diabetic conditions (hyperinsulinemia) in the STZ-nicotinamide treated Wistar rats. Despite high insulin levels in the diabetic rats, the blood glucose levels were enhanced implicating towards perturbed insulin signaling pathway. Conversely, Morin supplementation restored insulin levels up to 0.80-fold (p < 0.05) compared to diabetic rats alluding its insulin stabilizing effect (Fig. 6a). Insulin sensitization was assessed by monitoring random and fasting blood glucose levels in rats. Data demonstrate that Morin significantly lowered random blood glucose levels (0.80-fold; p < 0.05) compared to diabetic rats (Fig. 6b). Furthermore, glucose tolerance test revealed Morin-mediated enhanced regulation over glucose load under diabetic conditions (Fig. 6c). Hyperglycemic toxicity also affected the body weight of diabetic rats which was significantly reduced after 4 weeks of diabetes induction compared to control rats, whereas Morin treatment arrested the abnormal decline in body weight of diabetic rats (Fig. 6D). Metformin treatment also protected against hyperglycemia-induced depreciation in body weight of rats; however, it could not avert diabetes-induced renal damage as kidney/body weight ratio was not lowered significantly compared to diabetic rats (Fig. 6e). Interestingly, Morin treated diabetic group showed a decrease in kidney/body weight ratio (p < 0.05) compared to diabetic rats implicating its remarkable reno-protective capacity (Fig. 6e). Notably, Morin and metformin co-treated rats (Mo + Met) could avert renal damage up to 1.29-fold (p < 0.05) as compared to the diabetic group. Thus, the data demonstrate that a) type 2 diabetes/ insulin resistance triggered nephrotoxicity in rats and b) it was largely mitigated by Morin and Mo + Met treatment.

Morin prevents diabetes-induced metabolic aberration in rats. a Serum insulin levels were assessed by ELISA assay in control and treated rats. Bar graph denotes serum insulin levels in ng/mL. b Graph denotes random and fasting blood glucose levels (mg/dL) in control and treated rats after 4 weeks of diabetes induction. c Graph denotes blood glucose levels obtained at 30 min regular intervals of oral glucose treatment (2 g/Kg bwt.) in rats up to 2 h during OGTT test. d Graph shows mean body weight (g) change in the rats after 4 weeks of indicated treatments. e Kidney weight/body weight ratio was calculated to examine the extent of damage in kidneys. Renal toxicity markers were assessed in serum samples. f Graph denotes fold change of serum blood urea nitrogen (BUN) levels in treated rat samples. g Graph shows serum creatinine levels in treated rat samples. Data are presented as mean ± SE *p < 0.05 vs control, #p < 0.05 vs diabetic
STZ-nicotinamide-induced diabetes caused renal dysfunction in rats as observed by elevated levels of serum renal damage markers. Diabetes enhanced serum BUN (1.7-fold, p < 0.05) and creatinine (1.6-fold; p < 0.05) levels compared to control indicating the decline in renal function (Fig. 6f, g). Interestingly, treatment with Morin averted renal impairment by significantly lowering serum creatinine and BUN (blood urea nitrogen) levels (Fig. 6f, g). Anti-hyperglycemic effects of Morin were compared with the standard anti-diabetic drug, Metformin. As expected, metformin lowered creatinine (0.82-fold, p < 0.05) and BUN levels (0.58-fold, p < 0.05), compared to diabetic rats. However, profound improvement in renal impairment was reflected by Morin and (Mo + Met) treatment compared to Metformin only treatment (Fig. 6f, g) suggesting that Morin curbs Metformin-induced toxicity in rats.
Inactivation of PHLPP1 by Morin protects renal cells against hyperglycemic insult
Loss of PHLPP1-mediated Nrf2 action influenced the redox capacity of cells tremendously as observed through the excessive generation of reactive oxygen species and superoxide radicals (Fig. 7a). Hyperglycemic cells displayed a significant increase in DCF fluorescence (1.10-fold, p < 0.05) indicative of ROS formation. Morin treatment suppressed generation of free radical species as assessed by reduced fluorescence intensity (Fig. 7a). Upon specific estimation of superoxide radicals by DHE dye, an increase of 1.45-fold, p < 0.05 was observed in the DHE/ethidium fluorescence ratio of hyperglycemic cells compared to control, implicating compromised antioxidant defense system (Fig. 7b). Morin, on the other hand, quenched superoxide generation as DHE fluorescence reduced to 0.6-fold (p < 0.05) compared to hyperglycemic cells (Fig. 7b). Furthermore, as Nrf2 is widely known to regulate GCLC gene (Glutamate–Cysteine Ligase Catalytic subunit), a rate-limiting enzyme of GSH synthesis, we examined GSH levels using CMFDA fluoroprobe, which is converted to chloromethyl fluorescein by intracellular esterases and reacts with glutathione. CMF fluorescence intensity was found to be significantly reduced (0.42-fold; p < 0.05) in the hyperglycemic cells (Fig. 7c). However, treatment with Morin protected renal cells by preventing the GSH depletion, thereby enriching the antioxidant pool.

Morin quenches hyperglycemia-induced oxidative stress in vitro and in vivo. a Total reactive oxygen species (ROS) were examined by incubating NRK52E cells with 10 μM DCFHDA dye after treatment (48 h) with high glucose, Morin and Metformin. Graph shows mean DCF fluorescence intensity. b Superoxide levels were estimated by determining the DHE/ ethidium fluorescence ratio in renal cells treated with 10 μM DHE dye after completion of treatment (48 h) with high glucose, Morin and Metformin. c CMFDA fluorescence was assessed to inspect the level of glutathione in the cells. GSH derived antioxidant pool was assessed in control and treated NRK52E cells. d Lipid peroxidation was determined in the treated rat renal tissue lysates by measuring the thiobarbituric acid reactive substances (TBARS). The graph shows nM TBARS/mg protein. e Total thiol content was determined in the in vivo samples showing significant depreciation in the thiol levels during diabetes. f Bar graph represents fold change of control in thioredoxin reductase activity of treated rat samples. g Glutathione reductase (GR) activity was assessed in control and treated renal tissue lysates. Activity is expressed in fold change of control. h Nrf2-dependent catalase activity was evaluated in renal samples of treated rats. The bars represent fold change in catalase activity. Data are presented as the mean ± SE, *p < 0.05 vs corresponding control, #p < 0.05 vs hyperglycemia/diabetic
In addition, we estimated oxidative stress in the kidney tissue lysates, where lipid peroxidation was enhanced significantly by 2.0-fold (p < 0.05) in the diabetic renal samples compared to control. Anti-oxidative potency of Morin proficiently attenuated lipid peroxidation by reducing TBARS content (0.65-fold, p < 0.05; Fig. 7d). Co-treatment of Morin with Metformin (Mo + Met) revealed efficient repression of oxidative stress compared to metformin only treatment (Fig. 7d). Thiol levels are an indicator of the total reducing equivalents of cells, thus, we determined total thiol content in the kidney lysates using DTNB reagent (5,5-dithio-bis-2-nitrobenzoic acid). Free sulfhydryl groups were significantly reduced in diabetic rats by 0.8-fold (p < 0.05) compared to control rats confirming weak antioxidant defense under hyperglycemic stress. Morin accorded significant protection by preventing the loss of thiol content in the diabetic kidney (Fig. 7e). Activities of anti-oxidative enzymes, such as thioredoxin reductase and glutathione reductase, declined significantly up to 0.5 and 0.25-fold (p < 0.05), respectively, in diabetic tissue lysate compared to control rats (Fig. 7f, g). Nrf2-dependent catalase activity was also declined significantly (0.44-fold; p < 0.05) depicting suppressed Nrf2 activation during diabetes compared to control (Fig. 7h). Nrf2 inducible property of Morin was once again established as it significantly increased the catalase activity by 2.56-fold (p < 0.05) compared to diabetic rats (Fig. 7h). Altogether, the results show that aberrant metabolic and redox conditions prevailed due to suppressed Nrf2 activation via PHLPP1 which contributed towards renal injury.
Inhibition of PHLPP1 by Morin caused ablation in FoxO1-mediated renal cell death
PHLPP1 activation and concomitant oxidative stress generation-induced apoptosis in diabetic kidneys through induction of pro-apoptotic proteins. Immunoblotting data reveal that FoxO1 responsive apoptotic proteins including Bax (Bcl-2 associated X protein), Bim (Bcl-2-like protein 11) and Puma (p53 upregulated modulator of apoptosis) were enhanced significantly in the diabetic renal lysates compared to control (Fig. 8a). Impact of PHLPP1 on renal apoptosis was assessed via phosphorylation of PHLPP1-dependent pro-apoptotic protein, Mst1 (macrophage stimulating 1). Phosphorylation of Mst1 was declined by0.59-fold (p < 0.05) in the diabetic rats. Moreover, the expression of the anti-apoptotic protein, Bcl2 (B-cell lymphoma 2) was decreased significantly (0.83-fold, p < 0.05) compared to control indicating activation of apoptotic cascade during diabetes (Fig. 8a, b). Remarkably, Morin supplementation prevented Bax release and concomitantly upregulated Bcl2 expression which prevented cleavage and activation of caspase3 protein (Fig. 8a–-c). Interestingly, silencing of PHLPP1 decreased Bim expression significantly in NRK52E cells compared to the hyperglycemic cells as apparent by immunocytochemistry data (Fig. 8d). It was observed that hyperglycemia promoted FoxO1-mediated Bim expression (3.03-fold; p < 0.05), while PHLPP1 gene knockdown restricted Bim induction significantly (Fig. 8d). Morin significantly prevented Bim release demonstrating its PHLPP1 inhibitory potential (Fig. 8d). Similarly, Mst1 protein (induced by PHLPP1) was reduced significantly by Morin as revealed through immunocytochemical studies in NRK52E cells (Fig. 8e). PHLPP1 silenced cells induced Mst1 phosphorylation resulting in its inactivation. In addition, PI (propidium iodide) uptake assay was performed to validate apoptosis induction during hyperglycemia as it is readily permeabilized in apoptotic cells. Morin supplementation substantially reduced apoptotic cell death up to 0.6-fold (p < 0.05) compared to hyperglycemic cells (Fig. 8f). Altogether, the data implicate that supplementation of Morin or gene silencing of PHLPP1 could efficiently restrict FoxO1-mediated apoptosis during hyperglycemia. Results highlight the role of PHLPP1/ Nrf2–FoxO1 pathway as a major contributing factor for renal pathogenesis during diabetic nephropathy. Our findings indicate the therapeutic benefit of Morin in quenching PHLPP1/Nrf2-dependent cell death during diabetes. Nrf2 not only enhances the anti-oxidant function but also regulates the expression of Mdm2 consequently affecting FoxO1 stability. Our results provide convincing experimental evidence that Morin inhibits PHLPP1-induced FoxO1 activation and can be used for the management of metabolic disorder and prevent kidney damage during diabetes.

Hyperglycemia induces PHLPP1/FoxO1-mediated renal cell death. a Immunoblots of FoxO1 and PHLPP1 regulated apoptotic proteins in diabetic, Morin and/or metformin treated rat renal lysates. β-actin was used as a loading control. b, c Densitometry of the immunoblots shown in panel A. Data are presented as mean ± SE, *p < 0.05 vs control, #p < 0.05 vs diabetic. d Immunocytochemistry of Bim protein in high glucose and Morin treated NRK52E cells. Expression of Bim (FITC) was also checked by gene silencing of PHLPP1. The images were captured at 20X; scale bar 50 μm. e Images represent immunocytochemistry of Mst1 protein (Alexa Fluor) in high glucose and Morin treated NRK52E cells. Expression of Mst1 was also assessed after gene silencing of PHLPP1. f Bar graph depicts propidium iodide uptake in NRK52E cells. Fluorescence was captured at Ex/Em 535/610 nm. Data are presented as mean ± SE (n ≥ 3), *p < 0.05 vs control, #p < 0.05 vs hyperglycemia
Discussion
The present work expands our understanding of how apoptosis is triggered during hyperglycemia and elucidates the underlying molecular mechanism. This study shows that activation of PHLPP1 impairs Nrf2 signaling causing inhibition of Mdm2-mediated FoxO1 ubiquitination. Results further demonstrate that renal cells were protected from hyperglycemic apoptosis by Morin through PHLPP1 inhibition and concurrent potentiation of Nrf2-mediated Mdm2 expression. Although FoxO1-mediated apoptosis has been a subject of robust investigation, the mechanisms through which it promotes cell death responses are not well understood. Herein, we identified a novel mechanism, where diabetic renal apoptosis is regulated by PHLPP1/Nrf2/FoxO1 axis.
PHLPP proteins are amongst the most characterized Ser/Thr phosphatases that dephosphorylate in the hydrophobic motif of Akt, PKC and S6K in isoform specific manner [50]. PHLPP1 is highly specific for Akt2, an Akt isoform involved with the metabolic regulation [52]. Besides this, PHLPP phosphatases regulate pro-apoptotic kinase, Mst-1, which is regulated via inhibitory phosphorylation on Thr387 by Akt [53]. Upon its dephosphorylation, Mst1 becomes activated and phosphorylate its downstream targets; p38 and JNK to induce apoptosis and growth arrest [54]. Correspondingly, enhanced Mst1 levels in the hyperglycemic lysates were observed, while its phosphorylation was enhanced upon Morin supplementation implicating apoptotic inhibition (Fig. 8A). Mst1 also phosphorylates FoxO1 at Ser 212 to promote its nuclear activity [55]. Rationally, PHLPP activates the Mst1 signaling pathway, which induces downstream responses via pro-apoptotic MAPK pathway [54]. Thus, apoptotic effects of PHLPP1 are mediated not only through the inactivation of the PI3K/Akt pathway but additionally through the activation of Mst1 regulated pro-apoptotic pathways. This raised the possibility of PHLPP1 in advancing dysregulation of cell survival during hyperglycemia. Loss in insulin-mediated cell signaling (PI3K/Akt) relays its dephosphorylated substrates including FoxO1, to induce specific growth arrest genes. Thus, we set out to define a possible role for PHLPP1 in diabetic renal disease. In the present study, we adopted a two-way approach to understand the mechanistic details of PHLPP1, where we reasoned that a) upregulation of PHLPP1 may disrupt Akt-mediated FoxO1 phosphorylation and b) Nrf2/Mdm2 axis would potentially disrupt FoxO1 degradation to promote the pathogenesis during DN. To test this possibility, we examined levels of PHLPP1 and phospho-Akt in diabetic renal lysates and identified its impact on FoxO1 stability (Fig. 1). Consistent with depreciated FoxO1 phosphorylation due to enhanced PHLPP1 expression, we observed a modest correlation between Nrf2 and Mdm2 in suppressing FoxO1 degradation (Fig. 4). Interestingly, this correlation was lost in Morin treated renal cell lysates probably due to reinforced Nrf2 activity and lowered PHLPP1 activation (Fig. 3). Contrarily, loss in PHLPP1 expression by gene silencing substantially restored Akt activity, thus inactivating FoxO1 to alleviate diabetes progression (Fig. 2).
Much discussed ubiquitin E3 ligase Skp2 (subunit of the Skp1/Cul1/F-box protein ubiquitin complex is also reported to regulate the ubiquitination and degradation of FoxO1, but to date no mutational or conformational studies have been performed to show the direct involvement of Skp2 E3 activity in FoxO ubiquitination, whereas Mdm2 is considered as a general ubiquitin E3 ligase for all mammalian FoxO factors [10, 14]. Essentially, Mdm2 regulates the expression of insulin-like growth factor 1 receptor (IGF1R) [56], holding a promising target therapy in diabetes. Moreover, Mdm2 is proposed to be a survival factor for podocytes and a crucial molecule for maintenance of renal function during diabetes [57, 58]. Taken this into account, it further persuaded us to inspect FoxO1/Mdm2 axis during diabetic kidney disease (DKD). Indeed, a correlation of Nrf2-mediated Mdm2 induction and an in-turn regulation of PHLPP/Akt axis on Nrf2 stability add a larger significance to this study. Inhibiting PHLPP1 by genetic strategy confirmed the pivotal role of Mdm2 in mediating high glucose-induced renal death through events promoting FoxO1 nuclear translocation and suppressing oxidative stress. To identify if the signaling is entirely mediated downstream to Akt, we limited PI3K/Akt activity by treating renal cells with LY29002. The data showed that blocking of Akt phosphorylation exacerbated FoxO1 activity as confirmed through immunoblotting studies (Fig. 3). Furthermore, phosphorylation of Mdm2 was lowered with LY29002 treatment (Akt inhibition) similar to the hyperglycemic group validating the critical role of the Akt pathway in hyperglycemic cell death response.
Hyperglycemia promoted expression of FoxO1-dependent pro-apoptotic proteins including Bim and Puma (Fig. 8). Simultaneously, expression of Mst1 was also enhanced significantly due to PHLPP1/FoxO1 axis, while Morin suppressed it, preventing renal apoptosis. In addition, selective gene knockdown of PHLPP1 not only enhanced FoxO1 degradation but also reinforced Nrf2-dependent anti-oxidative defense system (Fig. 7). Consistently, in line with the in vitro findings, the STZ-Nicotinamide injected hyperglycemic rats also showed FoxO1 activation relating more towards the pathogenesis of DN. The effects of Morin were compared to that of metformin, where it was observed that metformin although conferred renal protection against hyperglycemic damages; however, the defensive potency and the extent of molecular modulatory capacity was subsidiary when compared to Morin. Enhancement of FoxO1 degradation by Morin indicated its insulin-sensitizing efficacy as previously reported (28,59) which was further verified by insulin ELISA assay, where Morin treatment restored the serum insulin levels compared to diabetic rats and corrected the glucose influx as monitored through glucose uptake assay (Fig. 5E). It is worth noting that metformin also regulated FoxO1 activity; however, the cell survival rate was higher with Morin supplementation.
The current study adds PHLPP1 as an important regulator that could be related to the pathogenic role of FoxO1 in DN. Taken together, PHLPP1 responses obstructs insulin signaling via PI3K/Akt–Nrf2 pathway. These signaling events were traced not only in vitro but also in vivo in the rat model of diabetes at early stages. Hyperglycemic milieu induces cooperation between these two pathways resulting in augmented oxidative stress and cell death. Data suggest that regulation of PHLP1/Nrf2/FoxO1 pathway may be a novel mechanism to avert diabetes-induced renal damages.
Conclusion
In conclusion, our findings indicate that elevated PHLPP1 levels play an important role in promoting FoxO1 nuclear stability during diabetes resulting in renal apoptotic cell death. We further observed that Morin, a bioflavonoid modulated PHLPP1/Nrf2 axis that prevented FoxO1-dependent apoptotic cell death during hyperglycemic stress. Morin also exhibited marked protection against STZ–nicotinamide-induced diabetic renal injury. Furthermore, our results identify that Morin could fortify Nrf2–ARE binding due to potential inhibition of PHLPP1. This not only restored the anti-oxidative capacity of the cell but also improved the glucose metabolic capacity of the cell through directly participating in the restoration of metabolic enzymes. Overall, the study demonstrates protective role of Morin in promoting anti-apoptotic effects through its molecular intervention in PHLPP1/Foxo1 axis during diabetic nephropathy.
Availability of data and material
Data will be available upon suitable request.
Abbreviations
- ARE:
-
Antioxidant response element
- AKT:
-
Akt murine thymoma viral oncogene
- BAX:
-
Bcl-2 associated X protein
- BCL2:
-
B-cell lymphoma 2
- BIM:
-
Bcl-2-like protein 11
- BUN:
-
Blood urea nitrogen
- Bwt.:
-
Body weight
- DN:
-
Diabetic nephropathy
- ELISA:
-
Enzyme-linked immunosorbent assay
- FoxO1:
-
Forkhead-box O1 protein
- G6P:
-
Glucose-6-phosphate
- G6K:
-
Glucose 6 kinase
- HG:
-
High glucose
- HO-1:
-
Hemeoxygenase 1
- IR:
-
Insulin resistance
- MAPK:
-
Mitogen activated protein kinase
- MDM2:
-
Mouse double minute 2 homolog
- MST1:
-
Mammalian sterile 20-like kinase 1
- NA:
-
Nicotinamide
- NEH:
-
Nrf2–ECH homology
- NQO1:
-
NAD(P)H dehydrogenase (quinone 1)
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- PIP3:
-
Phosphatidylinositol 3,4,5-triphosphate
- PKB:
-
Protein kinase B
- PHLPP1:
-
Pleckstrin homology domain and leucine rich repeat protein phosphatase 1
- PKC:
-
Protein kinase C
- PI3K:
-
Phosphoinositide 3-kinase
- PUMA:
-
P53 upregulated modulator of apoptosis
- STZ:
-
Streptozotocin
- T2D:
-
Type 2 diabetes
- Ub:
-
Ubiquitin
References
Reidy K, Kang HM, Hostetter T, Susztak K (2014) Molecular mechanisms of diabetic kidney disease. J Clin Investig 124(6):2333–2340
Jha JC, Banal C, Chow BS, Cooper ME, Jandeleit-Dahm K (2016) Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid Redox Signal 25(12):657–684. https://doi.org/10.1089/ars.2016.6664
Van Buren PN, Toto R (2011) Hypertension in diabetic nephropathy: epidemiology, mechanisms, and management. Adv Chronic Kidney Dis 18(1):28–41. https://doi.org/10.1053/j.ackd.2010.10.003
Fox CS, Matsushita K, Woodward M, Bilo HJ, Chalmers J, Heerspink HJL et al (2012) Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without diabetes: a meta-analysis. The Lancet 380(9854):1662–1673. https://doi.org/10.1016/S0140-6736(12)61350-6
Wilding JP (2014) The role of the kidneys in glucose homeostasis in type 2 diabetes: clinical implications and therapeutic significance through sodium glucose co-transporter 2 inhibitors. Metabolism 63(10):1228–1237. https://doi.org/10.1016/j.metabol.2014.06.018
Maiese K (2015) FoxO transcription factors and regenerative pathways in diabetes mellitus. Curr Neurovasc Res 12(4):404–413
Tzivion G, Dobson M, Ramakrishnan G (2011) FoxO transcription factors; Regulation by AKT and 14–3–3 proteins. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1813(11): 1938–1945. http://dx.doi.org/https://doi.org/10.1016/j.bbamcr.2011.06.002
Manning BD, Toker A (2017) AKT/PKB signaling: navigating the network. Cell 169(3):381–405. https://doi.org/10.1016/j.cell.2017.04.001
Boccitto M, Kalb R (2011) Regulation of Foxo-dependent transcription by post-translational modifications. Curr Drug Targets 12(9):1303–1310
Huang H, Tindall, DJ (2011) Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1813(11):1961–1964. http://dx.doi.org/https://doi.org/10.1016/j.bbamcr.2011.01.007
O’neill BT, Lee KY, Klaus K, Softic S, Krumpoch MT, et al (2016) Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J Clin Investig 126(9):3433–3446. https://doi.org/10.1172/JCI86522
Huang Y, Hu J, Zheng J, Li J, Wei T, Zheng Z, Chen Y (2012) Down-regulation of the PI3K/Akt signaling pathway and induction of apoptosis in CA46 Burkitt lymphoma cells by baicalin. J Exp Clin Cancer Res 31(1):48
You A, Nam CW, Wakabayashi N, Yamamoto M, Kensler TW, Kwak MK (2011) Transcription factor Nrf2 maintains the basal expression of Mdm2: an implication of the regulation of p53 signaling by Nrf2. Arch Biochem Biophys 507(2):356–364
Fu W, Ma Q, Chen L, Li P, Zhang M, Ramamoorthy S et al (2009) MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J Biol Chem 284(21):13987–14000. https://doi.org/10.1074/jbc.M901758200
Mathur A, Pandey VK, Kakkar P (2017) PHLPP: A putative cellular target during insulin resistance and type 2 diabetes. J Endocrinol 233(3):R185–R198
Mathur A, Pandey VK, Kakkar P (2018) Activation of GSK3β/β-TrCP axis via PHLPP1 exacerbates Nrf2 degradation leading to impairment in cell survival pathway during diabetic nephropathy. Free Radical Biol Med 120:414–424
Andreozzi F, Procopio C, Greco A, Mannino GC, Miele C, Raciti GA et al (2011) Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia 54(7):1879–1887. https://doi.org/10.1007/s00125-011-2116-6
Grzechnik AT, Newton AC (2016) PHLPPing through history: a decade in the life of PHLPP phosphatases. Biochem Soc Trans 44(6):1675–1682. https://doi.org/10.1042/BST20160170
Zhang Z, Liew CW, Handy DE, Zhang Y, Leopold JA, Hu J et al (2010) High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and β-cell apoptosis. FASEB J 24(5):1497–1505. https://doi.org/10.1096/fj.09-136572
Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA (2018) Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis 9(2):119. https://doi.org/10.1038/s41419-017-0135-z
Montgomery MK, Turner N (2015) Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect 4(1):R1–R15. https://doi.org/10.1530/EC-14-0092
Manda G, Checherita AI, Comanescu MV, Hinescu ME (2015) Redox signaling in diabetic nephropathy: hypertrophy versus death choices in mesangial cells and podocytes. Mediators Inflamm 604208:13. https://doi.org/10.1155/2015/604208
Rizvi F, Mathur A, Krishna S, Siddiqi MI, Kakkar P (2015) Suppression in PHLPP2 induction by morin promotes Nrf2-regulated cellular defenses against oxidative injury to primary rat hepatocytes. Redox Biol 6:587–598. https://doi.org/10.1016/j.redox.2015.10.002
Mathur A, Rizvi F, Kakkar P (2016) PHLPP2 down regulation influences nuclear Nrf2 stability via Akt-1/Gsk3β/Fyn kinase axis in acetaminophen induced oxidative renal toxicity: Protection accorded by morin. Food Chem Toxicol 89:19–31
Pandey VK, Mathur A, Kakkar P (2018) Emerging role of Unfolded Protein Response (UPR) mediated proteotoxic apoptosis in diabetes. Life Sci 216:246–258. https://doi.org/10.1016/j.lfs.2018.11.041
Pandey VK, Mathur A, Khan MF, Kakkar P (2019) Activation of PERK-eIF2α-ATF4 pathway contributes to diabetic hepatotoxicity: Attenuation of ER stress by Morin. Cellular signaling 12:41–52. https://doi.org/10.1016/j.cellsig.2019.03.008
Kapoor R, Kakkar P (2012) Protective role of morin, a flavonoid, against high glucose induced oxidative stress mediated apoptosis in primary rat hepatocytes. PLoS ONE 7(8):e41663. https://doi.org/10.1371/journal.pone.0041663
Paoli P, Cirri P, Caselli A, Ranaldi F, Bruschi G, Santi A et al (2013) The insulin-mimetic effect of Morin: a promising molecule in diabetes treatment. Biochimica et Biophysica Acta (BBA)-General Subjects 1830(4): 3102–3111. http://dx.doi.org/https://doi.org/10.1016/j.bbagen.2013.01.017
Li X, Kover KL, Heruth DP, Watkins DJ, Moore WV et al (2015) New insight into metformin action: regulation of ChREBP and FOXO1 activities in endothelial cells. Mol Endocrinol 29(8):1184–1194. https://doi.org/10.1210/ME.2015-1090
Song J, Ren P, Zhang L, Wang XL, Chen L, Shen YH (2010) Metformin reduces lipid accumulation in macrophages by inhibiting FOXO1-mediated transcription of fatty acid-binding protein. Biochem Biophys Res Commun 393(1):89–94. https://doi.org/10.1016/j.bbrc.2010.01.086
Zou J, Hong L, Luo C, Li Z, Zhu Y, Huang T et al (2016) Metformin inhibits estrogen-dependent endometrial cancer cell growth by activating the AMPK–FOXO 1 signal pathway. Cancer Sci 107(12):1806–1817. https://doi.org/10.1111/cas.13083
Samuel VT, Choi CS, Phillips TG, Romanelli AJ, Geisler JG, Bhanot S et al (2006) Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes 55(7):2042–2050. https://doi.org/10.2337/db05-0705
Nwadozi E, Roudier E, Rullman E, Tharmalingam S, Liu HY, Gustafsson T, Haas TL (2016) Endothelial FoxO proteins impair insulin sensitivity and restrain muscle angiogenesis in response to a high-fat diet. FASEB J 30(9):3039–3052
Masiello P, Broca C, Gross R, Roye M, Manteghetti M, Hillaire-Buys D, Ribes G (1998) Experimental NIDDM: development of a new model in adult rats administered streptozotocin and nicotinamide. Diabetes 47(2):224–229. https://doi.org/10.2337/diab.47.2.224
Kapoor R, Kakkar P (2014) Naringenin accords hepatoprotection from streptozotocin induced diabetes in vivo by modulating mitochondrial dysfunction and apoptotic signaling cascade. Toxicol Rep 1:569–581. https://doi.org/10.1016/j.toxrep.2014.08.002
Tahara A, Matsuyama-Yokono A, Nakano R, Someya Y, Shibasaki M (2008) Hypoglycaemic effects of antidiabetic drugs in streptozotocin-nicotinamide-induced mildly diabetic and streptozotocin-induced severely diabetic rats. Basic Clin Pharmacol Toxicol 103(6):560–568. https://doi.org/10.1111/j.1742-7843.2008.00321.x
Tsuji K, Taminato T, Usami M, Ishida H, Kitano N, Fukumoto H, Seino Y (1988) Characteristic features of insulin secretion in the streptozotocin-induced NIDDM rat model. Metabolism 37(11):1040–1044.40.
Lamont BJ, Visinoni S, Fam BC, Kebede M, Weinrich B, Papapostolou S et al (2006) Expression of human fructose-1, 6-bisphosphatase in the liver of transgenic mice results in increased glycerol gluconeogenesis. Endocrinology 147(6):2764–2772
Himmelfarb J, McMonagle E, McMenamin E (2000) Plasma protein thiol oxidation and carbonyl formation in chronic renal failure. Kidney Int 58(6):2571–2578
Niknahad H, Shuhendler A, Galati G, Siraki AG, Easson E, Poon R et al (2003) Modulating carbonyl cytotoxicity in intact rat hepatocytes by inhibiting carbonyl metabolizing enzymes. II Aromatic aldehydes Chemical Biology Interactions 143–144:119–128
Sinha AK (1972) Colorimetric assay of catalase. Anal Biochem 47:389–394. https://doi.org/10.1016/0003-2697(72)90132-7
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70–77
Mannervik B (1999) Measurement of Glutathione Reductase Activity. Current protocols in Toxicology, Chapter 7, Unit7.2.
Zhang J, Liu Y, Yang HW, Xu HY, Meng Y (2009) Molecular mechanism of beta cell apoptosis induced by p58 in high glucose medium. Sheng Li Xue Bao 61(4):379–385
Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vásquez-Vivar J, Kalyanaraman B (2003) Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radical Biol Med 34(11):1359–1368
Hedley DW, Chow S (1994) Evaluation of methods for measuring cellular glutathione content using flow cytometry. Cytometry 15:349–358. https://doi.org/10.1002/cyto.990150411
Hay N (2011) Interplay between FOXO, TOR, and Akt. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1813(11):1965–1970.
Mackenzie RW, Elliott BT (2014) Akt/PKB activation and insulin signaling: a novel insulin signaling pathway in the treatment of type 2 diabetes. Diabetes, metabolic syndrome and obesity: targets and therapy 7:55. https://doi.org/10.2147/DMSO.S48260
Sasaki M, Sasako T, Kubota N, Sakurai Y, Takamoto I, Kubota T et al (2017) Dual regulation of gluconeogenesis by insulin and glucose in the proximal tubules of the kidney. Diabetes 66(9):2339–2350. https://doi.org/10.2337/db16-1602
Brognard J, Newton AC (2008) PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol Metab 19(6):223–230
Newton AC, Trotman LC (2014) Turning off AKT: PHLPP as a drug target. Annu Rev Pharmacol Toxicol 54:537–558. https://doi.org/10.1146/annurev-pharmtox-011112-140338
Qiao M, Wang Y, Xu X, Lu J, Dong Y, Tao W et al (2010) Mst1 is an interacting protein that mediates PHLPPs’ induced apoptosis. Mol Cell 38(4):512–523. https://doi.org/10.1016/j.molcel.2010.03.017
Meng Z, Moroishi T, Guan KL (2016) Mechanisms of Hippo pathway regulation. Genes Dev 30(1):1–17. https://doi.org/10.1101/gad.274027.115
Yuan Z, Lehtinen MK, Merlo P, Villén J, Gygi S, Bonni A (2009) Regulation of neuronal cell death by MST1-FOXO1 signaling. J Biol Chem 284(17):11285–11292. https://doi.org/10.1074/jbc.M900461200
Warfel NA, Newton AC (2012) Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP): a new player in cell signaling. J Biol Chem 287(6):3610–3616. https://doi.org/10.1074/jbc.R111.318675
Worrall C, Suleymanova N, Crudden C, Drakensjö IT, Candrea E, Nedelcu D (2017) Unbalancing p53/Mdm2/IGF-1R axis by Mdm2 activation restrains the IGF-1-dependent invasive phenotype of skin melanoma. Oncogene 36(23):3274
Ye C, Tang H, Zhao Z, Lei CT, You CQ, Zhang J et al (2017) MDM2 mediates fibroblast activation and renal tubulointerstitial fibrosis via a p53-independent pathway. American Journal of Physiology-Renal Physiology 312(4):F760–F768
Lei CT, Tang H, Ye C, You CQ, Zhang J, Zhang CY et al (2017) MDM2 contributes to high glucose-induced glomerular mesangial cell proliferation and extracellular matrix accumulation via Notch1. Sci Rep 7(1):10393. https://doi.org/10.1038/s41598-017-10927-5
Naowaboot J, Wannasiri S, Pannangpetch P (2016) Morin attenuates hepatic insulin resistance in high-fat-diet-induced obese mice. J Physiol Biochem 72(2):269–280. https://doi.org/10.1007/s13105-016-0477-5
Acknowledgements
The authors thank CSIR-IITR review committee for evaluation of the manuscript and providing the manuscript communication no.3592.
Funding
This study was supported by grants from CSIR project (HCP-0019). Senior Research fellowships to AM, MFK, VKP from CSIR and UGC is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
The study was conceptualized and designed by AM and PK. Experimentation, analysis and data interpretation was done by AM, VKP and MFK. Manuscript drafting and writing work was a joint effort of AM, VKP and PK. All authors have discussed and contributed to the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no potential conflict of interest.
Ethics approval
All experiments on rats were conducted in accordance with the international guidelines and protocols approved by the Institutional Animal Ethics Committee (IAEC) of CSIR-IITR with approval no. IITR/IAEC/01/16–82/16.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mathur, A., Pandey, V.K., Khan, M.F. et al. PHLPP1/Nrf2–Mdm2 axis induces renal apoptosis via influencing nucleo-cytoplasmic shuttling of FoxO1 during diabetic nephropathy. Mol Cell Biochem 476, 3681–3699 (2021). https://doi.org/10.1007/s11010-021-04177-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-021-04177-3