
Abstract
Classifier chains are an effective technique for modeling label dependencies in multi-label classification. However, the method requires a fixed, static order of the labels. While in theory, any order is sufficient, in practice, this order has a substantial impact on the quality of the final prediction. Dynamic classifier chains denote the idea that for each instance to classify, the order in which the labels are predicted is dynamically chosen. The complexity of a naïve implementation of such an approach is prohibitive, because it would require to train a sequence of classifiers for every possible permutation of the labels. To tackle this problem efficiently, we propose a new approach based on random decision trees which can dynamically select the label ordering for each prediction. We show empirically that a dynamic selection of the next label improves over the use of a static ordering under an otherwise unchanged random decision tree model. In addition, we also demonstrate an alternative approach based on extreme gradient boosted trees, which allows for a more target-oriented training of dynamic classifier chains. Our results show that this variant outperforms random decision trees and other tree-based multi-label classification methods. More importantly, the dynamic selection strategy allows to considerably speed up training and prediction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Contrary to multi-class classification, where only one class label is expected to be associated to an example, multi-label classification (MLC) is the task of assigning a subset of all possible labels to an example (Tsoumakas and Katakis 2007). A well known example for such a problem is automatic text categorization where the goal is to assign a set of relevant categories to a document (Schapire and Singer 2000). Most of the current approaches address these problems with problem transformation methods where the problem is split up into multiple smaller subtasks which are solved independently. Binary Relevance (BR) (Joachims 1998; Godbole and Sarawagi 2004; Boutell et al. 2004) is the best known example, which decomposes the multi-label problem into a set of N independent binary classification problems, one for each label. However, this decomposition ignores possible dependencies between the labels.
Classifier chains (CC) solve this problem by learning one model for each label, but taking the predictions of the previous models along a predetermined sequence of the labels into account (Read et al. 2011, 2021). To this end, the models are arranged along a predefined chain where each model passes its own and all previously predicted labels on to the next model in the chain, which incorporates them as additional input features. It can be shown that CC are able to capture local as well as global dependencies, and that these are crucial for minimizing non-deomposable loss functions, which cannot be reduced to the marginal label errors (Dembczyński et al. 2012).
However, while these theoretical results are independent of the selected chain order, in practice the performance of CC highly depends on the order of the labels along the chain. There are several reasons for this, among them, e.g., the propagation of label errors through the chains: if the first label in the chain is incorrectly selected, this error may affect all subsequent predictions (Senge et al. 2014). Finding a good sequence is a non-trivial task because (i) the number of possible sequences to consider grows exponentially with the number of labels, and (ii) local dependencies may make it necessary to consider different chains for different instances. For example, it is arguable easier to detect first a car and then infer its headlights in an image taken at daylight, whereas it is easier to first detect the lights and from that deduce the car in a night image. Moreover, methods which try to explore different orderings are usually computationally expensive so that the most frequently selected option is to pick a random ordering.
The assumption in this work is that the ordering in which labels should be predicted in order to obtain the best performance highly depends on the specific context, namely the test instance at hand. Dynamic chaining addresses this problem of how to dynamically choose an appropriate ordering for individual instances instead of the entire datasets. The main problem that needs to be solved is that, if the label prediction order can be dynamically selected at prediction time, we need to prepare the classifier for an exponential number of potential label orderings. A naïve adaptation of CC for this setting, would therefore have to train up to N! different chains, which is clearly infeasible. The contribution of this work is to investigate tree-based ensembles for an efficient solution of this task.
Our first contribution is the adaptation of random decision trees (RDT) (Zhang et al. 2010) for the purpose of constructing dynamic chains. In contrast to the common induction of decision trees or to random forests, RDTs do not optimize a heuristic splitting criterion, but instead select the splits at the inner nodes randomly. We adapt RDTs to dynamic chains by allowing tests on the labels at the inner nodes, which can be turned on or off. This has the advantage that the objective can easily be changed during prediction without the need for modifying the trees. In an iterative process, the learned RDTs are repeatedly queried to determine the next most certain (positive or negative) label, which is then added to the input features, and the respective label tests in the RDTs are turned on. Furthermore they are fast to train and can achieve competitive and robust performance without optimizing any objective function (Zhang et al. 2015). Most importantly, RDTs allow us to embed static and dynamic chains in exactly the same trees, so that we can compare the two approaches in a controlled environment, with otherwise identical models. The results of this experimental evaluation confirm that dynamic classification chains clearly outperform static orderings.
However, despite the appealing simplicity and flexibility of RDT, this comes at the expense of predictive performance in comparison to other tree-based MLC models, since RDTs are not trained in order to optimize a particular measure. We therefore further adapt extreme gradient boosted trees (XGBoost) (Chen and Guestrin 2016), a highly optimized and efficient tree induction method, to the DCC setting. The resulting classifier, XDCC,Footnote 1 thus integrates DCC into the extreme boosting structure of gradient boosted trees. XDCC’s optimization goal in each boosting round is to predict only one single positive label for which it is the most certain. This label can be different for each training instance and depends on the given data, label dependencies, and previous predictions for the instance at hand. The information about the predicted labels is carried over to subsequent rounds. A key advantage of the proposed approach is the reduced run time resulting from the fact that we do not need to predict the entire chain of labels (N predictors), but only the actually relevant labels for each instance, which is typically much smaller (usually below 10). Hence, only few rounds are potentially enough if only the positive labels are predicted, whereas CC-based approaches have to still make predictions for each of the existing labels.
In summary, our contributions are the following:
-
We present a general motivation and a thorough formalization of dynamic classifier chains, and a brief review of previous work in this area (Sect. 3)
-
We show how to adapt random decision trees so that static and dynamic classifier chains can be modeled in the same structure, and thus be compared to each other within an otherwise identical model. The results confirm a clear advantage for dynamic over static classifier chains (Sect. 4).
-
We introduce a multi-label formulation of the XGBoost objective which is much more efficient than the decomposition based XGBoost baselines, and propose a variant of XGBoost which integrates dynamic classifier chains, yielding a versatile and efficient multi-label classifier (Sect. 5).
-
We demonstrate empirically that the XGBoost variant outperforms other tree- and ensemble-based multi-label classifiers, especially regarding the computational costs (Sect. 6).
This paper is based on (Kulessa and Loza Mencía 2018) and (Bohlender et al. 2020). It provides an expanded, unified and definite description of these works, puts a stronger emphasis on the DCC framework, a more complete discussion of related work, and more detailed as well as several new experimental results.
2 Multi-label classification
This section briefly recapitulates previous work in multi-label classification that is relevant for us and clarifies the notation used throughout this paper. Extensive overviews of MLC are provided, e.g., by Tsoumakas and Katakis (2007), Tsoumakas et al. (2010), or Zhang and Zhou (2014).
2.1 Problem definition and simple transformation methods
Multi-label classification is the task of learning a mapping from instances \(X \in {\mathcal {X}}\) to subsets of a finite set of non-exclusive class labels \({\mathcal {Y}}=\{y_1, \ldots , y_N\}\). Equivalently, it may be viewed as an instance of multi-target prediction (Waegeman et al. 2019), where the task is to predict for a finite set of N unique class labels \(\Lambda =\{\lambda _1,\ldots ,\lambda _N\}\) whether they are positive (or relevant) or negative (or irrelevant). Formally, \(y_j=1\) if \(\lambda _j\) is relevant, and \(y_j=0\) if \(\lambda _j\) is irrelevant for a given instance. Thus, the training set consists of training examples \(\mathrm {\mathbf {x}}_i \in X\) and associated label sets \(\mathrm {\mathbf {y}}_i \in {\mathcal {Y}}=\{0,1\}^N\), \(1 \le i \le M\), which can be represented as matrices \(X=(x_{iq}) \in A^{M\times Q}\) and \(Y=(y_{ij})\in \{0,1\}^{M\times N}\), where features \(x_{ij}\) can be represented as continuous, categorical or binary values. An MLC classifier \(f: {\mathcal {X}} \rightarrow {\mathcal {Y}}\) uses the training set in order to learn the mapping between input features and output labels. The prediction of f for a test example \(\mathrm {\mathbf {x}}\) is a binary vector \({{\hat{\mathrm {\mathbf {y}}}}}=f(\mathrm {\mathbf {x}})\).
The simplest solution to MLC is binary relevance decomposition (BR), where each label is treated as an independent classification task for which a classifier is trained. Formally, we learn a function \(f_i: {\mathcal {X}} \rightarrow \{0,1\}\) for each different \(\mathrm {\mathbf {y}}_i\) that has been observed in the training data. As a result, each prediction for a label is independent of the predictions of the other labels, which is the main disadvantage of this simple technique.
At the other end of the spectrum is the label power set transformation (LP), which reduces the problem of MLC to a single multi-class classification task, where each possible label combination is encoded as a separate and exclusive class. By predicting all labels at once, this approach naturally takes label dependencies into account. In addition to the obvious limitations due to the exponential growth of label combinations, LP does not allow to predict label combinations which have not been seen in the training data. However, this may also be viewed as an advantage under certain circumstances (Senge et al. 2014).
2.2 Classifier chains
Classifier chains (Read et al. 2011, 2021) overcome the disadvantages of the above-mentioned approaches, because they neither assume full label independence nor full dependence. As in BR, a set of N binary classifiers is trained, but in order to being able to consider dependencies, the classifiers are connected along a chain. Each classifier takes the predictions of all previous ones as additional features in order to predict the respective label. More specifically, each \(f_j\) is trained on an augmented training set \(X^{(j)} = [X,Y_{\cdot ,1},\ldots , Y_{\cdot ,j-1}]\) containing the true labels in order to predict the j-th target \(Y_{\cdot ,j}\) of Y. At prediction time, \(f_j\) predicts \(\hat{y_j}\) based on previous predictions \(\hat{y_1},\ldots ,{\hat{y}}_{j-1}\), i.e., \({\hat{y}}_j= f_j(\mathrm {\mathbf {x}},\hat{y_1},\ldots ,{\hat{y}}_{j-1})\) with \({\hat{y}}_1=f_1(\mathrm {\mathbf {x}})\).
Dembczyński et al. (2010) analyzed classifier chains in a probabilistic setting, in which the joint probability of the labels can be estimated via the Bayesian chain rule. In theory, this results in Bayes optimal predictions, independent of the chosen order of the labels. However, in practice, the resulting probabilistic classifier chains (PCC) have a much higher time complexity for finding the label combination with the maximum joint probability, and are thus only feasible for datasets with no more than 15 labels (Dembczyński et al. 2010). To tackle this problem beam search (Kumar et al. 2013) or A* search (Mena et al. 2015) can be used to perform the inferences, which significantly speeds up the process for determining the most probable label subset. Mena et al. (2016) give an overview of inference methods for PCC. Nevertheless, PCC also rely on a predefined, static chain ordering.
Further research revealed that CC and PCC are able to capture global dependencies as well as dependencies appearing only locally in the instance space (Dembczyński et al. 2012). However, while the decomposition is, in theory, order independent according to the Bayesian chain rule, in practice, the performance of classifier chains depends on the chosen, static ordering of the labels (da Silva et al. 2014). Consequently, a variety of techniques have been proposed which aim at determining a good static chain sequence in advance. For this purpose methods such as genetic algorithms (Goncalves et al. 2013), prior statistical analyses and Bayesian networks (Malerba et al. 1997; Sucar et al. 2014), ordering according the difficulty of the single-label problems (Kumar et al. 2013), formulating it as dynamic programming problem (Liu and Tsang 2015), or a double Monte Carlo optimization technique (Read et al. 2014) have been proposed. Alternatively, Read et al. (2011) already suggested to form an ensemble of different chains, each corresponding to a different, randomly selected permutation of the labels, a proposal that was later refined by Li and Zhou (2013).
However, creating and maintaining an ensemble of CCs is not always feasible (Goncalves et al. 2013) and also poses the non-trivial problem of combining multiple, dependent multi-label predictions (Nguyen et al. 2020). More importantly, static label ordering techniques which use the previous predictions to estimate the next label can practically only tackle dependencies in one direction, which may not be optimal for making predictions. Indeed, already Malerba et al. (1997) found that projecting label dependencies into a sequential ordering is a non-trivial task. Moreover, especially for tasks with local dependencies, which differ in different parts of the instance space, a static ordering may only be able to capture half of the exploitable dependencies in the worst case. Taking into consideration such dependency structures require a dynamic, example-dependent approach of ordering the predictions. We will return to this question in Sect. 4.
2.3 Evaluation measures
A large variety of evaluation measures have been proposed in MLC, which differ, e.g., in the importance they attribute to label dependencies. For our purposes, the most interesting ones are Hamming accuracy, subset accuracy, and the F1 measure.
Hamming accuracy (HA) denotes the accuracy of predicting individual labels averaged over all labels.
where \(\mathbb {I}\) denotes the indicator function. As each label is evaluated independently of the others, binary relevance methods typically perform quite well.
Subset accuracy (SA), on the other hand, measures the ability of a classifiers to predict exactly the target label set.
Thus, methods such as the label power set approach can be expected to perform comparatively well on this measure. However, if the number of labels is rather large, subset accuracy is often of limited use since most of the predictions will be wrong in at least one label, causing SA to evaluate to zero.
Hence, we additionally consider example-based F1 (F1) to measure the performance. Rooted in information retrieval, its key idea is to evaluate the harmonic mean between the precision (how many of the predicted labels are relevant?) and the recall (how many of the relevant labels have been predicted?) of the predicted labels for each example, averaged over all examples. F1 can be considered as a compromise between HA and SA.
The measures also differ in the computational complexity they incur: in order to minimize SA, we must find the mode of the joint label distribution, whereas it is sufficient to find the modes of the marginal label distributions for HA. If there are dependencies between labels, both modes do not have to coincide. Hence, the trade-off between both measures and the relation to BR can serve to assess the ability of considering label dependencies.
As the objective of classifier chains is to find the correct label combination, we expect the impact of our proposed extensions to be best reflected in SA. The F1 measure is less extreme than SA, since it also considers partial matches and is therefore often used for providing a general comparison of the predictive quality. Though it is sufficient to obtain good estimates for the individual labels in order to optimize univariate losses such as F-measure or Hamming loss (Dembczyński et al. 2012), this is not necessarily the case when there are dependencies between the labels.
2.4 Datasets
By now, there are many benchmark datasets for multi-label classification available, which cover a wide variety of application areas.Footnote 2 From these, we selected the datasets shown in Table 1, which have various characteristics in terms of the number of instances and labels, the average cardinality of the relevant labels per example, and the number of distinct label sets that occur in the training data. Not visible from the statistics (and generally not known) are correlations and dependencies between the labels. As we have discussed above, chaining approaches can only be expected to gain an advantage over, e.g., BR if there are global or local dependencies between the labels in the dataset, which can be picked up and modeled by the learner. For instance, there is evidence that yeast and enron contain mostly global dependencies whereas scene also exhibits local dependencies (Papagiannopoulou et al. 2015; Loza Mencía and Janssen 2016; Moyano et al. 2017). Unfortunately, so far only few works have tried to systematically analyze these characteristics. All datasets came with predefined train-tests splits which were used for the evaluation.
3 Dynamic classifier chains
Classifier chains depend on a fixed, static ordering of the labels. As we have discussed above (Sect. 2.2), this problem is typically tackled by a heuristic choice of a suitable ordering, by pooling the result of multiple orderings, or by simply selecting a random ordering. Apart from the computational disadvantages of exploring different label sequences, the underlying assumption that there is one unique, globally optimal ordering which fits equally to all instances, can also be questioned. It is therefore natural to investigate whether a suitable classifier chain can be chosen depending on the test instance at hand.
In this section, we will first motivate the need for dynamic classifier chains (Sect. 3.1), then formalize the problem (Sect. 3.2), and finally review prior work in this area (Sect. 3.3), before we introduce tree-based solutions to this problem in the following Sects. 4 and 5.
3.1 Motivation

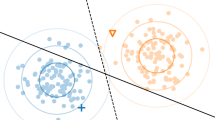
Example for different classifications using the static and the dynamic ordering for a two different pictures. See text for explanations
Consider a hypothetical example of labeling an image with all objects that appear in it. More specifically, we want to identify cars and their parts in their surroundings. Figure 1 shows two example images, which both show a car, one taken by day in a forest, and one by night in a city. Classifier chains would predict the possible labels on both pictures in a static, heuristically or randomly selected order. For the sake of the example (and without loss of generality), we show an alphabetical ordering in the first row below the pictures. It is first predicted whether the scenery shows a beach or not, then whether it shows a car, a forest, a headlamp, a road, and so on. Each prediction depends on the previous ones (and on the input image itself). It seems natural to assume that a good order would predict the label for which it is most certain about first. Moreover, as a prediction for a label \(\lambda _i\) influences the predictions of all subsequent labels \(\lambda _j, j>i\), predicting the most certain labels first will also help to minimize the effects of error propagation along the chain.
However, clearly, if we try to sort the labels according to the certainty with which they can be recognized in each of the pictures, we obtain a different chain in each of the images. For example, whereas predicting the presence of a headlamp can benefit from the previous rather simple detection of a car in the left picture, the opposite is the case in the dark picture on the right where the headlamps are considerably easier to detect than the car itself. While the static chain of alphabetically ordered labels in the CC model depicted in the first row can only exploit the local dependency on the left picture, the dynamic approach illustrated in the second row, which predicts first the labels for which it is most certain, can also take advantage from the rather easy detection of headlamp on the right by leaving the prediction of car for later.
Note, however, that many of the certain predictions are actually negative. For example, we may quite reliably infer that the left pictures does not show a beach, or that the right picture does not show a forest. In many applications, such predictions are not desirable. So even though they might be helpful, it feels more natural to have a chain that contains only positive dependencies. Moreover, it is certainly more efficient, particularly considering that in MLC the number of positive labels usually stays in the tens even when the total number of labels is into the hundreds or thousands. The approach in the last row chooses first the labels for which it is most certain that they are present. After the predictions of forest and road, respectively, we can already stop the detection process since only negative predictions are to come. The advantage comes at the expense of ignoring dependencies to negative labels. Predicting the absence of a label is often much easier than finding positive ones and the knowledge about the absence of a label might be very useful to predict a positive label. For instance, the detection of forest may benefit from the information that beach is not in the scenery of the left picture, as used by the second approach.
In the remainder of the paper, we will discuss tree-based approaches that are able to learn dynamic prediction chains, and, in one case, also focus on positive labels only. But first, we will formally define the problem.
3.2 Problem definition
From a formal point of view, adapting the order of the predicted labels simply corresponds to a context-dependent re-ordering or the chain rule. More specifically, we can represent the joint distribution as
where \(\pi\) is a permutation over N in one-line notation, i.e., \(\{\pi _k \, | \,1 \le k \le N\}=\{1,\ldots ,N\}\).
CC estimates the mode of \(P(\mathrm {\mathbf {y}}\, | \,\mathrm {\mathbf {x}})\) by greedily maximizing \(f_k(\mathrm {\mathbf {x}},{\hat{y}}_{\pi _1},\ldots ,{\hat{y}}_{\pi _{k-1}}) \approx P(y_{\pi _k} \, | \,y_{\pi _1}, \ldots , y_{\pi _{k-1}}, \mathrm {\mathbf {x}})\) using a fixed, predetermined \(\pi\). A dynamic approach, instead, determines \(\pi\) depending on the instance \(\mathrm {\mathbf {x}}\) at hand. For instance, labels could be ordered according to certainty, i.e., the closeness of the conditional probability to 0 or 1:
as in the second setting depicted in Fig. 1. Ordering according to descending probability, as for the model in the last row, corresponds to choosing \(\pi\) as
which would order the chain rule for an instance with p positive labels as (\(\pi (\mathrm {\mathbf {x}})\) written as \(\pi\) for convenience)
Recall that theoretically, by the product chain rule, all decompositions are equivalent and independent of the chosen permutation (Dembczyński et al. 2010), i.e.,
for two different permutations \(\pi\) and \(\pi '\) of the label set. However, in practice, the corresponding probability estimates and the resulting classifiers \(f_k(\mathrm {\mathbf {x}}, {\hat{y}}_{\pi _1} \dots {\hat{y}}_{\pi _{k-1}})\) are error prone, in which case the order of the labelings does matter. To see this, assume a simple problem where for each example either all labels are present (\(\forall j: y_j = 1\)), or all labels are absent (\(\forall j: y_j = 0\)). However, each of the labels is noisy, so that the BR classifiers \(f_j(\mathrm {\mathbf {x}})\) have different error rates \(\epsilon _j\). Without loss of generality, let \(\epsilon _1< \epsilon _2< \dots < \epsilon _N\). The HA (1) of the binary relevance classifier is the average accuracy \(\frac{1}{N}\sum _j(1 - \epsilon _j)\). Let us further assume that these errors are correlated, so that a classifier chain is able to pick up the signal that all subsequent classifiers just repeat the first label, i.e., \(f_k(\mathrm {\mathbf {x}}, {\hat{y}}_{\pi _1} \dots {\hat{y}}_{\pi _{k-1}}) = {\hat{y}}_{\pi _1}\) for all \(k > 1\). The accuracy of CC now clearly depends only on the error of the first member in the chain \({\hat{y}}_{\pi _1} = f_{\pi _{k-1}}(\mathrm {\mathbf {x}})\), i.e., on \(\epsilon _{\pi _1}\). If CC is trained using identity permutation (\(\pi _k = k\)), its HA will be \(1-\epsilon _1\) and therefore higher than the one for BR. If CC is trained on the inverse permutation (\(\pi _k = N+1-k\)), its HA will be \(1-\epsilon _N\), and therefore lower than BR. The expected HA for a randomly selected chain order will be the same as the HA for BR.
So, in the above simple example, the performance of CC, and whether it is able to outperform BR, clearly depends on the chosen label ordering, and, coincidentally, the frequent default choices of selecting a random label order, or of averaging among several different orders, will, in expectation, not outperform BR.
If we now slightly modify the above example, so that label errors \(\epsilon _j\) are not evenly distributed, but that certain labels can be better predicted in some regions of the example space, and worse in other regions, we can, by essentially the same argumentation, arrive at the conclusion that an optimal classifier for the above problem needs to identify the most reliable label for a given test example, and start the chain with this label. This is essentially the idea of the approaches that we will discuss in the remainder of the paper.
3.3 Related work
The idea that the optimal order in which labels are predicted may depend on the input example at hand is not entirely new, and has been tried in various settings before. Da Silva et al. (2014) made a first attempt by letting a nearest neighbor classifier decide which ordering to use for a given instance. However, the dynamic selection was restricted to a pre-determined set of static label orderings. It is also computationally expensive since new CC models have to be build during prediction. Nam et al. (2017) use recurrent neural networks to predict the positive labels as a sequence, but the ordering in which these are predicted is pre-determined. Nam et al. (2019) attempted to solve this problem using reinforcement learning and recurrent neural networks, but found that this does not outperform simple ordering strategies. Moreover, the employed reinforcement learning approach essentially comes down again to exploring many possible label sequences during training, which is computationally very demanding. Llerena and Deratani Mauá (2017) propose several approaches for MLC based on sum-product networks, including a sequential classification method which can either use a static or a dynamic ordering, which focuses on predicting positive labels first. However, the authors do not examine the difference between these two strategies. Trajdos and Kurzynski (2019) introduce dynamic chaining based on accuracy estimates of binary relevance predictions in a local neighborhood of the test example, and employ this technique for nearest neighbor and Naïve Bayes classifiers, but, in the latter case, have to make a conditional independence assumption on the label predictions as well.
4 Dynamic classifier chains with random decision trees
In this section, we show how dynamic classifier chains can be efficiently constructed using an extension of RDTs, which are particularly appealing for this purpose: First, as we will see in the following, we can collapse BR and other MLC transformation or decomposition methods (Tsoumakas et al. 2010) such as CC to a single RDT ensemble without loss in predictive accuracy, therefore saving memory and computational costs. Second, and more importantly, RDTs can provide a controlled environment where we can compare alternative decomposition methods, prediction methods and other extensions isolated from any side effects since the model can be fixed beforehand and be the same for every analyzed approach.
4.1 Random decision trees for multi-label classification
Fan et al. (2003) introduced RDTs as an ensemble of randomly created decision trees, i.e., as trees for which the tests at the inner nodes are chosen randomly during the construction. This is the major difference compared to classical decision tree algorithms, but also to the well known algorithm family of random forests (Breiman 2001), where only the subset of features which each tree learner can use is randomly drawn for each node. In contrast, RDTs do not optimize any objective function during training, yet they are able to achieve competitive and robust performance (Zhang et al. 2015). They were previously already successfully applied to MLC, focusing on large scale problems (Zhang et al. 2010) and streaming data with concept drift (Kong and Yu 2011).
4.1.1 Training
Starting from the root node, inner nodes of a random tree are constructed recursively by distributing the training instances according to the randomly chosen test at the inner node as long as the stopping criterion of maximum depth or minimum number of instances is not fulfilled. Discrete features are chosen without replacement for the tests in contrast to continuous features, for which additionally a randomly picked instance determines the threshold (Fan et al. 2005). In case that no further tests can be created, a leaf will be constructed in which information about the assigned instances will be collected. For MLC, we track the number of instances \(N_{v}^\theta\) in leaf v of tree \(\theta\) in relation to the number of positive values \(n_{v}^\theta (j)\) for label \(\lambda _j\).
4.1.2 Prediction
During prediction, an instance is forwarded from the root to a leaf node passing the respective tests in the inner nodes. In case some of the tested features have missing values, all branches are visited and the function \(q(\mathrm {\mathbf {x}},\theta ) \subset \{1,\ldots ,|\theta |\}\) returns a set of the leaf indices in tree \(\theta\) to which the instance has been assigned to. Following Fan et al. (2005), the posterior probability that the specific label \(y_{j}\) is true given an instance \(\mathrm {\mathbf {x}}\) and a tree \(\theta\) can be formalized as
As the randomness results in a large variety of distribution in an ensemble of RDT, many of them approaching the prior label distribution, we propose to distinguish between the quality of the collected statistics and to reward trees with higher confidences in their estimates. The Gini index is often used for determining the purity of a distribution, which we use in inverted form as follows
in order to weight the estimates of the individual trees, resulting in the overall prediction for the ensemble \(\Theta\) as
An obvious option in order to obtain multi-label predictions from the estimations in (11) is to use a threshold of \(50\%\) so that \({{\hat{y}}}_j = \mathbb {I}\left[ f_j(\mathrm {\mathbf {x}}) \ge 0.5\right]\). However, as Quevedo et al. (2012) observed, a threshold of 50% is not always ideal. Note that the tests in the tree are not specifically chosen to obtain a high purity of the distributions in the leaves, and in fact many leaves might contribute only with estimates close to the prior distribution, pulling down the average estimates. Thus, we follow Zhang et al. (2010) and estimate the average number of relevant labels as
where
\(R(\mathrm {\mathbf {x}},\Theta )\) is rounded in order to get an integer. This value is then used to cut the ranking of labels induced by the distribution of the marginals \(P(y_j=1 \, | \,\mathrm {\mathbf {x}}, \Theta )\).
4.2 Static chain ordering
The use of RDTs allows us to collapse a classifier chain to a single RDT in the following way: Instead of training N RDTs on the N augmented spaces \(X^{(k)} = [X,Y_{\cdot ,\pi _1},\ldots , Y_{\cdot ,\pi _{k-1}}]\), we train only one RDT on the complete augmented space \([X,Y] \in {\mathcal {X}} \times {\mathcal {Y}}\). This type of RDT can answer any query in the form of
by creating a query instance \((\mathrm {\mathbf {x}},\mathrm {\mathbf {p}})\) where the \(p_{\pi _1}=b_1, \dots , p_{\pi _{k-1}}=b_{k-1}\) are filled with the available label values, and all remaining \(p_j\) set as unknown or missing. The RDT can then answer the query by combining all possible paths for the missing values, as described in Sect. 4.1.2. A classifier chain prediction for a fixed label ordering \(\pi\) is hence obtained by initializing \(\mathrm {\mathbf {p}}^0\) with missing values and filling up \(p^k_{\pi _k}\) with the label predictions \({\hat{y}}_{\pi _k}=\mathbb {I}\left[ f_{\pi _k}(\mathrm {\mathbf {x}},\mathrm {\mathbf {p}}^{k-1})\right] \ge 0.5]\) while proceeding through the chain.
As RDTs are completely randomized, we can expect on average the same predictions as for a RDT ensemble which skipped the respective feature during training. In fact, in our experiments, we control the percentage of activated label tests with a parameter \(\sigma\), which allows us to analyze the effect of using previous predictions on an otherwise unchanged model.
Figure 2 visualizes the prediction process for a label on a single tree: Let us assume that the label to be predicted is \(y_i\), which comes before \(y_j\). In this case neither \(y_i\) nor \(y_j\) are known, i.e. all three colored branches are followed and the respective leaves are used in order to produce a prediction for \(y_i\). For label \(y_j\) the previous label \(y_i\) would be known, so that—depending on its value—we would skip either the left or the right branch, obtaining a label distribution at the leaves which is different and more refined than the previous one. Indeed, we can observe that the number of leaves on which the prediction relies, is monotonically decreasing during the classification process. Therefore, the set of leaves to which the instance is assigned in the first iteration will always be a superset of the leaves of the following iterations. This leads to a refinement of the predictions throughout the iterations.

Example for the refinement of a prediction for a particular instance and decision tree. \(y_i\) and \(y_j\) indicate tests on labels at the respective inner nodes
4.3 Dynamic chain ordering
In order to take advantage of the situation that predicting a label before or after another one might be easier depending on the instance at hand, we propose to let the RDT decide which label to predict next. Hence, instead of using the estimated probabilities to decide whether the i-th label in \(\pi\) is positive or negative, we use it to set the label for which RDT is most confident in its prediction. Labels, which were already predicted, are ignored.
Starting with the empty prediction vector \(\mathrm {\mathbf {p}}^0\), in each iteration i we select the label for which the RDT is most confident following (5):
Let us consider again the tree in Fig. 2. The difference to the static chain approach is that the aggregated blue, red and green leaves would be used in order to determine whatever label \(y_k\) is most likely given the found distribution, instead of a specific label (in the previous example label \(y_i\)). Hence, the RDT could decide to predict \(y_j\) instead if they are more confident about it, or any other label with the highest confidence.
The process of predicting the value needs further adaption due to the iterative prediction of the labels. The idea is to have predicted exactly \(R((\mathrm {\mathbf {x}},\mathrm {\mathbf {p}}^N),\Theta )\) positive labels after the prediction sequence is completed. Since the prediction \(\mathrm {\mathbf {p}}\) is constantly changed during the classification process, \(R((\mathrm {\mathbf {x}},\mathrm {\mathbf {p}}^k),\Theta )\) has to be re-computed in every iteration. First of all, we can only predict a label in iteration k positive if the number of already predicted positive labels \(|\mathrm {\mathbf {p}}^{k-1}|\) is smaller than \(R((\mathrm {\mathbf {x}},\mathrm {\mathbf {p}}^{k-1}),\Theta )\). Moreover, we have to predict a label as positive if we know that all the remaining labels in the chain need to be predicted positive to ensure that we obtain exactly \(R((\mathrm {\mathbf {x}},\mathrm {\mathbf {p}}^{k-1}),\Theta )\) positive labels. Hence, the k-th label is predicted as
4.4 Evaluation
A key aspect in our experimental evaluation was to verify our ideas of dynamic classifier chains on the usage of RDT as a controlled experimental environment for fair and specific comparisons. In particular, the focus was to demonstrate that using dynamic, context-dependent predictions improves over using static orderings w.r.t. predictive performance (Sect. 4.4.1). A decisive role in this is played by the influence of the previous predictions on the current prediction, which is analyzed in Sect. 4.4.2. Lastly in Sect. 4.4.3, we inspect the dynamic sequences in detail by evaluating the predicted labels in each iteration.
Unless otherwise noted, we have chosen to evaluate the parameter setting of using 300 decision trees, a maximum depth of 30, a maximum leaf size of 5 and a percentage of label tests of \(30\%\). Preliminary experiments with RDT revealed reasonable and stable performance for this parameter setting also on other kind of problems.
4.4.1 Static vs. dynamic label orderings
In this experiment we evaluated the advantage of the dynamic chain ordering in comparison to using a static chain ordering. Taking advantage of our controlled environment, we built for both approaches the same ensemble of trees, respectively. The only difference between the dynamic and the static setup is the ordering of the labels during the prediction process. We compare our proposed dynamic method to the averages over ten randomly-drawn but fixed orderings used for the static CC approach in Table 2.
The first and foremost observation is that the dynamic chain ordering is clearly superior to the static chain ordering on all datasets. This confirms our main hypothesis that it is advantageous to adapt the prediction order according to the context at hand. In fact, on most datasets the results for SA and F1 often doubles by using the dynamic chain instead of the static orderings. However, with respect to HA we can observe that only minor improvements can be achieved on the sparse datasets, that is, enron, medical, genbase, bibtex and tmc2007.

Influence of label tests on DCC. The y-axis represents the value for the measure and the x-axis represents the percentage \(\sigma\) of activated label tests. The color indicates the percentage of label tests per tree
4.4.2 Independent predictions vs. exploiting previous predictions
In this experiment we evaluated how the prediction is influenced by the usage of the label tests, i.e., by the usage of the previous predictions in the dynamic chain. At this stage the flexibility of the RDT algorithm pays off since we can choose the ratio \(\sigma\) of activated tests on the labels without the need for adaptations of the model (cf. Sect. 4.2). Hence, \(\sigma =0\) corresponds to a binary relevance classifier using RDT (more specifically, the collapsed version). Incrementing \(\sigma\) allows to directly observe utility and the effectiveness of exploiting potential label dependencies. Furthermore, we directly control the probability of choosing a test on a label feature at the inner nodes (10, 20 and 30).
Figure 3 shows the benefit for some selected datasets w.r.t. all evaluation measures (visualizations for all other datasets can be found in Fig. 11 in the appendix). For instance, we can observe on datasets emotions and yeast a major influence of the activated label tests on the performance. It seems obvious that there is a strong dependency between the labels in the datasets of which we can take advantage. The positive effect is less pronounced on some of the other datasets, depending on the measure and the label test configuration. For instance, we can observe a decrease in F1 for growing number of activated tests for enron especially for the 30% label test configuration. However, the ability of predicting the correct label combination (SA) does not seem to suffer.
4.4.3 Analysis of the dynamic sequences
Our approach dynamically produces a different prediction sequence on the labels for each given test instance. We were interested in characterizing and analyzing these sequences, which were selected by the RDT as being most appropriate for producing accurate predictions.
Figures 4 and 5 visualize our results exemplarily for flags and emotions. The heat map on the left shows the average accuracy (color) of predicting the j-th label in the dynamic sequence (y-axis) for different parameter configurations (x-axis), whereas the right map visualizes the number of labels (color) which were predicted as positive until a certain iteration.
We can observe on flags and emotions, as well as on the remaining datasets, that independently of the parameter configuration the predictions of the first iterations are much more accurate than the predictions at the end. One reason for this picture is, of course, that our label selection method specifically chooses the labels where the RDT ensemble is most confident first. This is also reflected by the heat maps on the right. They show that RDTs tend to first predict the (easier) negative labels before heading to the (more difficult) positives ones. Apparently, collecting as much as possible of the more readily accessible evidence helps to take the harder decisions later in the chain, as the comparison to the static chains in Sect. 4.4.1 demonstrates. This is the case even though later predictions may suffer more from error propagation.

Heatmaps characterizing the predicted sequences on flags

Heatmaps characterizing the predicted sequences on emotions
4.5 Discussion
Our experimental results based on the controlled evaluation environment of RDTs show that dynamic orderings improve over static orderings. In particular, the dynamic approach takes advantage by first predicting labels for which it is the most certain, which are then used to refine the estimates of the remaining, more challenging, labels in the following iterations.
With respect to computational complexity, the costs for building the trees and performing the dynamic predictions is essentially the same as for a static ordering. They mainly depend on the size of the ensemble and the depth of the trees. Moreover, the dynamic approach potentially allows to shorten the prediction process, namely when enough positive (or negative) labels have been already predicted, removing the dependencies on the label size.
However, as preliminary experiments with RDT have shown this strategy was consistently inferior to selecting according to the certainty. The reason might be again the non-optimized construction of the trees which leads to underestimated probabilities close to the prior that are difficult to differentiate and select from. An additional disadvantage due to the lack of any optimization is the potential performance gap to state-of-the-art methods. For instance, our results so far and additional comparisons to state-of-the-art approaches (cf. Sect. 6.4) show that RDTs are inherently not suitable for sparse data like text. To address this problem, we propose in the next section to replace the simple and flexible training process of RDTs by a tree induction method which specifically optimizes the dynamic chain prediction.
5 Learning a dynamic chain of boosted tree classifiers
As shown in the previous section, choosing the order of the labels dependent on the instance at hand can lead to a significant improvement in predictive accuracy, and dynamic classifier chains are able to exploit this. An additional potential advantage of DCC originates from the fact that multi-label problems may include a large number of labels, but the actual number of assigned labels to instances almost always stays low. As argued in Sect. 3, in such cases focusing on the positive labels could lead to a massive reduction in computational costs. For instance, in a MLC dataset with 100 labels but maximum number of assigned labels of 5, restricting the length of the chain to this number could be sufficient for the classification problem whereas a CC would still have to train 100 models and perform 100 classifications per instance. However, the advantage comes at the expense of not being able to consider dependencies to negative labels. In order to be still able to exploit the computational advantage, we need more reliable and targeted predictions for the positive labels than the ones that RDT could provide in our experimental results.
The highly optimized gradient boosting framework (Friedman 2002) is well suited for this task since it allows to maximize arbitrary differentiable objective functions. This is achieved by adding decision trees to an ensemble which are optimized in a gradient descent like procedure. Furthermore, its iterative procedure seems adequate in order to naturally integrate the dynamic chaining process. Similarly to DCC, gradient boosted trees are constructed by iteratively adding trees which refine previous predictions in a step-wise manner. The proposed XDCC approach integrates DCC into the state-of-the-art extreme boosting architecture of XGBoost (Sect. 5.3). As a preparatory step, we propose the extension ML-XGB of XGBoost which is able to perform multi-label instead of only binary predictions at the leaves (Sect. 5.2).
5.1 Extreme gradient boosted trees
Extreme Gradient Boosted Trees (XGBoost; Chen and Guestrin 2016) is a versatile implementation of gradient boosted trees. One of the reasons for its success is the very good scalability due to the specific usage of advanced techniques for dealing with large scale data. XGBoost was originally designed for dealing with regression problems, but different objectives can be defined by correspondingly adapting the objective function and the interpretation of the numeric estimates. Each model consists of a predefined number of decision trees. These trees are built using gradient boosting, i.e., the model is step-wise adding trees which further minimize the training loss. The main difference to RDT tree construction is the way the feature splits inside the nodes are determined. RDT uses completely random split tests, whereas XGBoost aims for finding splits that maximizes a gain score for the resulting leaves. The trees are constructed recursively, starting at the root node, by adding feature tests on the inner nodes. At each inner node, all possible feature tests are evaluated according to the gain obtained by applying the split on the data. The test candidate returning the highest gain score is then taken and both children are further split up until the maximum depth is reached or the gains stay below a certain threshold. A prediction can be calculated by passing an instance through all trees and summing up their respective leaf scores.
5.1.1 Boosted optimization
We refer to (Chen and Guestrin 2016) for a more detailed description of XGBoost. An XGBoost model consists of a sequence \(\theta ^{(1)}, \ldots , \theta ^{(T)}, T=|\Theta |\) of decision trees. Each tree \(\theta ^t\) returns a numeric estimate \(f^{(t)}(\mathrm {\mathbf {x}})\) for a given instance \(\mathrm {\mathbf {x}}\). Predictions are generated by passing an instance through all trees and summing up their leaf scores. The model is trained in an additive manner and each boosting round adds a new tree that improves the model most. For the t-th tree the loss to minimize becomes
where \({\hat{y}}_i^{(t-1)}=\sum _{t'=1}^{t-1} f^{(t')}(\mathrm {\mathbf {x}}_i)\) is the prediction of the tree ensemble so far, \(l(y,{\hat{y}})\) is the loss function for each individual prediction and \(\Omega\) is an additional term to regularize the tree. Combined with a convex differential loss function the objective can be simplified by a second-order approximation. This comes down to the following objective which can be minimized recursively at each node
where \(|\theta |\) is the size of the tree in number of leafs, \(G_v\) defines the sum of the gradients for all instances \(I_v\) in leaf v, \(H_v\) is the corresponding sum of the Hessians (cf. also Sect. 5.2), and \(\epsilon\) and \(\gamma\) are regularization terms derived from \(\Omega\). In such a leaf, the predicted score \(w_v^* = -\frac{G_v}{H_v + \epsilon }\) minimizes \({obj}^*\). The following gain function is used to evaluate different splits at inner nodes, where u and v for G and H refer to the resulting left and right leafs.
5.2 Multi-label XGBoost
Since XGBoost only supports binary classification with its trees in the original implementation, the underlying tree structure had to be adapted in order to support multi-label targets.
The first modification is to calculate leaf weights and gradients over all class labels instead of only a single one. More specifically, \(G_{j,v} = \sum _{i \in I_v} g_{j,i}\) and \(H_{j,v} = \sum _{i \in I_v} h_{j,i}\) extend to the labels \(1\le j \le N\), abbreviated as \(G_{j}\) and \(H_{j}\) for convenience. In consequence, the objective (18) and gain functions (19) have to be adapted to consider gradient and hessian values from all classes. A common approach in multi-variate regression and multi-target classification is to compute the average loss of the model over all targets (Waegeman et al. 2019). Adapted to our XGBoost trees, this corresponds to the sum of \(\frac{G_j^2}{H_j+\epsilon }\) over all labels (cf. Table 3). We refer to it as the sumGain split method. We use cross entropy as our loss, as it has demonstrated to be appropriate practically and also theoretically for binary and especially multi-label classification tasks (Nam et al. 2014; Dembczyński et al. 2012). Hence, the loss is computed as (shown here only for a single label)
In order to get \({\hat{y}}\) as a probability between zero and one, a sigmoid transformations has to be applied to the summed up raw leaf predictions \({\tilde{y}}=\sum _{t=1}^{T} f_t(\mathrm {\mathbf {x}})\), returned from all boosting trees, where \({\hat{y}} = \mathop {\mathrm {sigmoid}}\limits ({\tilde{y}}) = \frac{1}{1+e^{-{\tilde{y}}}}\). This is also beneficial for calculating g and h, since the gradients of the loss function simply become
One might not expect a very different prediction from the combined formulation than from minimizing the loss for each label separately by separate models (as by BR). However, as Waegeman et al. (2019) note, fitting one model to optimize the average label loss has a regularization effect that stabilizes the predictions, especially for infrequent labels. In addition, only one model has to be inferred in comparison to N, which has a major implication on the computational costs. This is especially an advantage in the case of a large number of labels and our proposed dynamic approach can directly benefit from it.
There are only few special adaptations of the gradient boosting approach to MLC in the literature and they mainly deal with computational costs. Both Si et al. (2017) and Zhang and Jung (2019) propose to exploit the sparse label structure which they try to transfer to the gradient and Hessian matrix by using L0 regularization. These approaches are limited to decomposable evaluation measures (such as (20)), which roughly speaking means that, opposed to the classifier chains approaches, they are tailored towards predicting the labels separately rather than jointly. Moreover, different technical improvements regarding parallelization and approximate split finding are proposed which could also be applied to the proposed technique in the following. Recently, Rapp et al. (2020) proposed to use gradient boosting in order to induce classification rules. Instead of predicting the labels in sequence, the rules predict all labels at once, which allows for minimizing also non-decomposable losses. On the other hand, previous predictions can only be exploited indirectly.
5.3 Gradient boosted dynamic classifier chains
After introducing the ML-XGBoost models, which can deal with multiple labels, the next step is to modify the tree construction to align it with our goal of predicting in each round a single label per instance. Depending on the strategy of ordering the labels, different ways of constructing the tree might be necessary. In our case, we adapt the tree construction process to the label ordering strategy by modifying the splitting criterion at the inner nodes.
Table 3 shows the proposed split functions and an example for each one to demonstrate the calculations. They replace the formulation of \({obj}^*\) in (19). In the example in the table, we assume to have a single instance with four different target labels \(\mathrm {\mathbf {y}}\in [0,1]^4\) and their corresponding predictions \({\hat{\mathrm {\mathbf {y}}}}\). g and h are calculated according to (21) and we get \(G=(-0.2,-0.8,0.9,0.1)\). Hereinafter we give a more detailed description and motivation for each gain function:
-
Maximum default gain over all labels
XDCC predicts labels one by one. It hence does not need to find a split which increases the expected loss over all labels (such as sumGain), but only one. Hence, maxGain is tailored to find the label with maximal gain, which corresponds to the label for which the previous trees produced the largest error. In the example in Table 3, this corresponds to \(\lambda _3\) for which a change of \(0.9^2\) w.r.t. cross entropy was computed if the prediction is changed to the correct one.
-
Sum and maximum gradients over all labels
In contrast to maxGain, sumSigned aims at good predictions for positive labels only and hence corresponds to the idea of predicting the positive labels first. Positive labels obtain positive scores, whereas negative labels obtain negative scores. The variant maxSigned chooses the positive label for which the greatest improvement is possible and only goes for the best performing negative label if there are no true positive labels in the instance set. In the example, \(\lambda _2\) is chosen since the improvement is greater than for \(\lambda _1\), and definitely greater as for the negative labels.
-
Sum and maximum absolute gradients over all labels
Different to sumGain and maxGain, the measures sumSigned and maxSigned not only favour positive labels but also take the gradients linearly instead of quadratically into account. This might, for instance, reduce the sensitivity to outliers. Hence, we also include two variants sumAbsG and maxAbsG which encourage to predict the labels where the model would improve the most, regardless whether it is positive and negative, but which similarly to sumSigned and maxSigned use a linear scale on the gradients.
Even though DCC’s original design is to predict a single label per round, good overall predictions might be required from the beginning for instance in the case of shorter chains. Therefore, we use the split-method as an additional hyperparameter to choose it individually for different XDCC variants and datasets.

Dynamic Chain: Example training pipeline (blue arrows) and prediction pipeline (red arrows) for a chain with length two that can predict up to two positive labels per instance
5.3.1 Training process
As for classical classifier chains, and differently from RDT, XGB trains a separate classifier for each label prediction round. The main difference to CC is of course again, that the next label to be predicted can be different for each instance, as it is chosen based on a dynamic prediction strategy. A schematic view for training the dynamic chain with a length of two is shown in Fig. 6 following the blue lines.
Similarly to RDT dynamic chain method in Sect. 4.2, we initialize the augmented label features \(\mathrm {\mathbf {p}}\) of each train instance \(\mathrm {\mathbf {x}}\) with “?".
While proceeding through the chain, these “?" values are replaced with predicted label probabilities out of prediction vector \({\hat{\mathrm {\mathbf {y}}}}^k \in (0,1)^N\). As soon as these feature columns begin to be filled with values, following classifiers may detect dependencies and base their predictions on them. In each round k, for \(1\le k \le N\), starts with training a new ML-XGBoost \(f^{1,k}\ldots f^{T,k}\) model of T trees by passing the train set combined with the additional label-features \(\mathrm {\mathbf {p}}^{k-1}\) and the target label matrix \(\mathrm {\mathbf {y}}\) to it. Afterwards, the model is used to generate predictions \({\hat{\mathrm {\mathbf {y}}}}^k=\mathop {\mathrm {sigmoid}}\limits \left( \sum _{t=1}^{T} f^{t,k}((\mathrm {\mathbf {x}},\mathrm {\mathbf {p}}^{k-1}))\right)\) on the same data used to train it, shown in the predictions tables. In the last step these predictions are then propagated to the next chain classifier by replacing the corresponding label features \(\pi _k\) chosen by the chaining strategy with the corresponding predicted probabilities, i.e., \(p^k_{\pi _k}={\hat{y}}^k_{\pi _k}\).
5.3.2 Dynamic chain ordering
In order to be able to shorten the training and prediction process we follow (6) and propagate the label with the highest estimated probability first. However, we combine this ordering on the positive labels with RDT’s strategy of selecting labels by their certainty (5) on the negative labels. More specifically, we start to select the label with the lowest probability next as soon as no further label with probability higher than 0.5 is found. The objective of this strategy is two-fold: in case the prediction process stops before round N, it is desirable 1) to have returned as many positive labels as possible and 2) that the predictions so far are as accurate as possible.
The strategy can be formalized as follows where \(J=\{1 \ldots N\}\backslash \{\pi _1 \ldots \pi _{k-1}\}\) denotes the labels which were not propagated previously:
Note that in practice it can still happen that a positive labels is found after a negative one, for instance because the negative predictions added evidence for a certain label to be relevant. For the same reason certainties for already set labels in \(\pi\) might also increase in subsequent rounds. In order to benefit from these increased certainties, we allow to update the scores in \(\mathrm {\mathbf {p}}\) in these cases. However, we do not allow that later classifiers revoke previous decisions by changing labels from positive to negative or the other way around.
5.3.3 Prediction process
The prediction process is similar to the training process. Instead of training a model in each step, we reuse the models from the training phase to generate predictions on the test set. After all predictions are propagated, the propagated labels are mapped to label predictions, where probabilities \(p_{j} < 0.5\) or equal to “?" are interpreted as negative labels and probabilities \(p_{j} \ge 0.5\) as positive labels. The process is depicted in Fig. 6 following the red lines.
5.3.4 Separate and conquer
We faced the following problem during the adaptation of the DCC approach to XGBoost. Consecutive models in the chain tend to select the same splits and therefore predict the same labels, especially ones which are easy to learn, e.g. if they clone existing features. We solve this problem by introducing an approach similar to separate-and-conquer from rule learning (Fürnkranz 1999) that is applied after each feature column update, i.e., after learning \(f^{1,k}\ldots f^{T,k}\) and as preparation for learning \(f^{1,k+1}\ldots f^{T,k+1}\). The separating step turns all gradient and hessian values of previously predicted labels for an instance to zero. Thereby, they are no longer considered during split score calculation in the conquering step and other splits become more likely since scores for already used splits are lower. The computation of the prediction scores \(w_v\) at the leafs is not affected by this measure so that labels still get the chance to be selected as next label for instances for which they were not yet chosen. This aspect is also relevant for the following measure.
5.3.5 Cumulated predictions
A second observation during development was that final predictions, after traversing the chain, contain too little positive labels. Analyzing the chain models showed that especially early models predict multiple positive labels, but are only allowed to propagate the one with the highest probability. Therefore we introduce cumulated predictions to preserve these otherwise forgotten positive predictions.
The idea is to save all predictions of each chain classifier and merge them afterwards with the chain predictions of the unmodified DCC using the following heuristic.
The final cumulated prediction \(c_{j}\) for label \(\lambda _j\) and an instance \(\mathrm {\mathbf {x}}\) is computed as
These final predictions \(c_{j}\) are used as in Sect. 5.3.3 in order to determine whether the label is set or not. For example, the standard version predicts \(\lambda _1\) as negative for test instance \(\mathrm {\mathbf {x}}_4\) in Fig. 6 since the label was chosen as next label in the first round and set to negative due to its probability of \({\hat{y}}^1_1=0.21\). In contrast, the cumulative approach would set \(\lambda _1\) as relevant since the second chain model predicted a probability of \({\hat{y}}^2_1=0.51\).
6 Experiments
The purpose of the experimental evaluation is two-fold. Firstly, we want to directly compare the proposed dynamic extension of gradient boosting trees to the static classifier chain variant, both in terms of predictive performance and computational costs (Sects. 6.2, 6.3). Secondly, we present a comparison to established decision tree learners, including the random decision trees proposed above, in order to assess the practical implications of the proposed extensions (Sect. 6.4). In particular, we evaluated the following algorithms:
-
J48: WEKA’s implementation of C4.5 represents in our comparison the family of classical single decision tree learners.
-
BR: Binary relevance learning, which learns on J48 decision tree for each label.
-
CC: Classifier Chains, which extend BR by including previously predicted labels as additional features for subsequent labels.
-
LP: The label powerset algorithm, which treats every label combination as a separate class value for J48.
-
RF: Random forest (and its variant predictive clustering trees) regularly achieve best positions in comparisons of state-of-the-art algorithms for multi-label classification (Madjarov et al. 2012; Bogatinovski et al. 2021, no comparison to gradient boosted trees, though). We used the WEKA implementation of random forests as base learner for BR, CC and LP.
-
XGB: XGBoost used as base learners for BR and CC.
-
RDT-DCC: Dynamic classifier chains using random decision trees (Sect. 4).
-
ML-XGB: A single multi-label XGBoost model introduced in Sect. 5.2.
-
XDCC\(_{\hbox {cum}}\): DCC with ML-XGB models as base classifiers, as proposed in Sect. 5.3, and cumulated predictions turned on.
-
XDCC\(_{\hbox {std}}\): The variant of XDCC without cumulated predictions, included in order to show the effect of this modification.
Hyper-parameters, especially regarding the tree construction, were optimized for F1 on a randomly selected 20% subset of the training set which was fixed beforehand.Footnote 3 This setting provided more stable results than using subset accuracy as the objective measure especially for the larger datasets.

Comparison with respect to length of the chain on yeast. XDCC-Order is overlayed by Rand-Order for F1. The shaded area for CC depicts the 5-quantiles (highest value, 20th, 40th, 60th, 80th value, lowest value)
6.1 Comparison of split functions
In a first step, we compared the proposed split gain functions. The average ranks in each column in Table 4 were obtained by optimizing hyper-parameters for the respective loss.Footnote 4
Our expectation was that the max heuristics, which finds splits leading to confident predictions for only one single label, should work particularly well with the proposed dynamic approach, since it fits to the idea of predicting labels one by one. In fact, the results show that the maxGain, maxSigned and maxAbsG methods generally beat their sum counter-parts in direct comparison. In particular maxGain, which is based on the idea of predicting the most confident labels first, is the best method (though the better methods are generally close together) also when considering the standard and cumulative version of XDCC separately. The maxSigned criterion, which tries to fulfill the requirement of predicting positive labels first, performs worse. However, the direct comparison between both methods w.r.t. F1 and XDCC\(_{\hbox {cum}}\), where maxGain won on 7 and maxSigned on 4 datasets, suggests that the best criterion depends to a great extent on the task at hand. Therefore, we included the selection of the appropriate splitting criterion as an additional hyper-parameter to be optimized in the following experiments.
The maxAbsG variants, which were meant to be less sensitive to outliers in the gradient computation, performed quite comparable to the base maxSigned variant. The bottom ranks of the sumSigned heuristic, especially in comparison to the much better sumAbsG variant, suggest that outliers are less problematic if the split is targeted to separate one single label where a confident prediction is possible.
6.2 Static vs. dynamic label orderings
Similarly to Sect. 4.4, we performed experiments which allow to see the advance of the DCC approach obtained by subsequently refining its predictions. Note, however, that contrary to the evaluation w.r.t. RDTs we cannot fully isolate the comparison between static and dynamic predictions from other effects than the order of the chain.
Hence, we included an instantiation of CC’s static chain which was generated by combining the dynamic chains found by XDCC for the instances in the validation set (XDCC Order). This ensures a certain proximity between the static and dynamic models and hence further cancels out, to a certain degree, effects caused by the random selection of the static orderings. Moreover, we included static but randomly ordered chains (Rand Order), and the static orderings from rare to frequent labels (Rare Order) and vice-versa (Freq Oder) as proposed by Nam et al. (2017). Each model used its own best hyper-parameters for the comparison of the predictive performance. However, XDCC used the XGBoost parameters found for CC for the comparison of the computational costs in order to obtain tree models of similar size.
As described in Sect. 5.3, XDCC can provide useful predictions after each round. This allows to terminate the prediction process early, which can be a major advantage over CC in terms of computational costs. Moreover, by subsequently refining its predictions based on previous predictions, we did not only expect to advance regarding F1, for which we optimized the XGBoost parameters, but especially in terms of SA. Figure 7 shows measures HA, SA and the time for training for different lengths of the chain in full detail exemplarily on yeast. The previous experiments on RDT (Sect. 4.4) showed that this dataset is appropriate to investigate the effect of static vs. dynamic label orderings, as it has a relatively high cardinality and appears to possess a label dependence structure which can be exploited by chaining techniques. The relatively small size of the dataset also allowed us to repeat CC 100 times in order to show the range of results if the permutations are chosen purely randomly. The trade-off between predictive performance and computational costs for the remaining datasets is analyzed further below. Note that length one of XDCC\(_{cum}\) corresponds to ML-XGB when the same parameter were used.

Train time ratios between XDCC\(_{cum}\) and CC in relation to their ratio with respect to F1 for nine datasets. Both models use individual XGB parameters where they performed best. CAL500 around (0.1,1.4) not shown for convenience, bibtex continues to (5.3,0.84).
The first observation in the two graphs in the top row is that, as expected, in terms of HA and SA, the performance of standard XDCC, which makes one prediction per label, improves with increasing length until a little bit further than the average cardinality of four of the dataset. If we add the cumulated predictions, the performances converge much faster because we can make more predictions in each iteration. Yet, there is a clear improvement visible for SA, which indicates that XDCC\(_{cum}\) is able to directly benefit from the previous predictions in order to match the correct label combinations. The cumulated predictions are also decisive for surpassing the static CC chains. This includes the 100 random ordering as well as the orderings according to the label frequencies and XDCC’s ordering. Interestingly, as can be seen from the red curves in the lower right graph, the training costs of CC are never reached although the same XGBoost parameters were used. In terms of prediction costs, XDCC becomes more expensive after eight rounds (green curve), which is long after it has reached the average cardinality and XDCC’s optimal predictive performance.

Train time ratios between XDCC\(_{cum}\) and CC in relation to their ratio with respect to F1 for nine datasets. Both models use the same XGB parameter set which was optimized for CC and F1. CAL500 starting at (0.008, 4.683) and ending (0.854, 4.690) not shown for convenience, no results for bibtex.
The point where train times of CC are reached by XDCC are further investigated in Figs. 8 and 9. It shows the ratio of XDCC\(_{cum}\) to CC for the different datasets (connected lines) and chain lengths. The difference between the two diagrams lies in the parameters used: XDCC was trained with the best parameters found for CC in Fig. 9. In contrast, CC and XDCC had separate hyper-parameter optimizations in Fig. 8. Hence, Fig. 9 is better suited for comparing the training times, whereas Fig. 8 allows for a better comparison of the reachable predictive performance. No clear general advantage was observed for neither of the frequency based label order heuristics in the previous analysis, especially not in comparison with a random order. Hence, we adopted static random chain orders in the following experiments. Note that optimizing a fixed chain, especially by trying out several different orderings, would multiply the training time by the number of different orders tried and thus be even more costly (cf. Sect. 2.2).
We can observe in both diagrams that XDCC is always faster in the first rounds than CC. Only for higher number of rounds the ratio is advantageous for CC, but at that point XDCC has always already converged w.r.t. predictive performance and additional rounds do not have a great impact. As already seen in Fig. 7 for yeast, we can also observe for the other datasets a steep increase in F1 in the first rounds which decelerates approximately when reaching the average number of positive labels per example. Consequently, the graphs for scene, birds, genbase and medical with a cardinality around one are straight or only exhibit an increase in the very beginning. Interestingly, the datasets for which XDCC has greater difficulties w.r.t. F1 are also the ones where XDCC has to invest more time than CC to learn the complete sequence. Except for CAL500, medical, emotions, the ordering of the endpoints seems to correlate quite well with the density of the datasets, i.e., the cardinality divided by the total number of labels. We leave further investigations of this relation for future work.

Heat maps of the development of the predictions of positive and negative labels (left and right side of the bar, respectively) from the first (top row) to last round (bottom row) given as fraction (color level) of the total number of positive and negative predictions on the respective dataset
The progress of predicting the labels is also depicted in Fig. 10. The fast completion of the prediction of positive labels, as visualized by the achievement of 100% on the respective left sides of the bars, indicates that positive labels are generally predicted in earlier rounds, as expected from the design of the split functions. As shown previously, this behaviour is decisive for the fast convergence and hence the possibility to end the training and prediction processes already in early rounds.
In summary, on the analyzed datasets, XDCC’s label ordering strategy allows the prediction process to terminate advance, resulting in a substantial speed-up in comparison to CC, without any major loss in predictive performance.
6.3 Comparison to decomposition methods
Table 5 summarizes the comparison between XDCC\(_{cum}\) and CC (random chain order) but also includes a binary relevance model trained with XGBoost and the other XDCC variants. Except for ML-XGB, which corresponds to stopping XDCC\(_{cum}\) after the first round, the XDCC models processed the full chain for both training and prediction. XDCC\(_{std}\) is included for showing the effect of cumulative predictions.Footnote 5
The first observation is the strong baseline achieved by BR regarding HA, as partially expected from Sect. 2.3. In the same way, CC is best in terms of SA. However, ML-XGB performs second regarding HA and XDCC\(_{cum}\) is second regarding SA, which suggests that the proposed approach is able to trade-off between both extremes. This is also confirmed by the best position in terms of F1. The positive effect of the cumulative predictions is clearly visible by the direct comparison between both XDCC. The comparison of the training and prediction times suggests that a similar advantage to BR is achievable as to CC when shortening the prediction process.
6.4 Comparison to baselines
Table 6 presents a comparison of the proposed tree-based dynamic classifier chain approaches to baselines based on the J48 tree learner and random forests. As different technical infrastructure are employed, we do not include comparisons of the computational costs. tmc2007 did not complete for J48 and RF on time and was therefore excluded from the comparison. Classifier chains used random static orderings.Footnote 6
Regarding Hamming accuracy, XDCC\(_{cum}\) achieved the highest average rank even though it is not targeted at making correct individual label predictions, as the comparison to the BR decomposition using XGBoost demonstrated previously. The more advanced techniques of gradient boosting seem to play out their advantage in this case. XDCC\(_{cum}\) also achieves good results on SA, though the label powerset method of RF clearly outperform the remaining algorithms on this measure. Senge et al. (2014) already showed that the LP method might be quite strong when only a small fraction of the \(2^N\) possible label combinations are observed in practice (or when the absolute number is generally low). They argue that approaches like BR or CC have to make up valid combinations by concatenating single decisions whereas LP can stick to combinations for which there is certainly evidence. Though these single decisions might be better than for LP, as seen in terms of Hamming accuracy, the probability that the full combination is valid decreases exponentially with N. Five of our dataset contain less than 100 distinct label combinations, and 7 less than 200, which might explain the good performance of LP. The advantage is taken over to F1, where RF-LP shares the first position with our proposed XDCC\(_{cum}\), but not to HA. As aforementioned, the RDT has difficulties regarding sparse dataset, which is the reason for the low performance in direct comparison to the more broadly purposed baselines. However, RDT-DCC surprisingly beats most of the baselines for F1.
More detailed results with raw performance scores for all the approaches and measures can be found in Tables 7, 8 and 9 in the appendix.
7 Conclusions
In this paper, we have shown that the static order of labels is a severe disadvantage of chain-based multi-label classifiers, and have proposed tree-based solutions to overcome this problem. This is achieved by dynamically selecting the next label in the sequence depending on the context, namely the instance at hand and the previously predicted labels for it. In comparison to other approaches for classifier chains, which have to learn appropriate sequences at training time, our first proposed approach comes at no additional cost, since the framework of random decision trees allows to perform the necessary inferences during prediction time. This also allowed us to confirm the importance of the dynamic label ordering on different datasets in a controlled setting, where identical random decision tree models were used for static and dynamic chain predictions, so that the observed advantage for the latter can be exclusively attributed to the dynamic label selection.
We have further proposed XDCC, an adaptation of extreme gradient boosted trees to dynamic classifier chains. It was shown that the positive labels are predominantly predicted at the beginning of the process, which allows XDCC to achieve its maximum performance already after a few rounds. This allows XDCC to reduce the length of the chain, which together with the multi-target formulation of XDCC leads to substantial performance improvements in comparison to binary relevance and classifier chains. The length of the chain also trades off between the two orthogonal objectives of binary relevance and classifier chains, leading to in average the best results in terms of F1.
A key limitation of our approach is that although the above results show that the process reaches optimal performance after a few iterations, we have not thoroughly investigated stopping criteria that would allow an early termination of that process. To that end, we plan to include a virtual label which indicates the end of the training and prediction process, similar to the idea of the calibrating label in pairwise learning (Loza Mencía et al. 2010). This will also help us to address problems with very large number of labels, which can be further facilitated by integrating some of the sparse techniques proposed by Si et al. (2017) and Zhang and Jung (2019). Since the number of associated labels per instance is usually not affected by the increasing number of labels, it will be interesting to see how XDCC will behave with respect to computational costs, since the size of the (dependency) chains should not grow significantly. Furthermore, we plan to transfer our ideas on dynamic chains to other kinds of algorithms, such as predictive clustering trees (Vens et al. 2008). Like random decision trees, their construction also does not depend on a specific target and is efficient, but they employ clustering which might yield more discriminative distributions at the leaves.
Data availability
The datasets we used can be found at http://mulan.sf.net/datasets-mlc.html and http://www.uco.es/kdis/mllresources/.
Code availability
The code of the XDCC algorithm is available at https://github.com/keelm/XDCC.
Notes
Publicly available at https://github.com/keelm/XDCC.
Repositories of multi-label datasets can be found at http://mulan.sf.net/datasets-mlc.html and http://www.uco.es/kdis/mllresources/.
The following parameters were tuned by grid-search: Number of trees {100, 300, 500}, max. tree depth {5, 10, 30, 50} for RF; Number of trees {100, 300, 500}, max. leaf size {3, 5, 9}, max. tree depth {5, 10, 20, 30, 50}, percentage of label tests {0.1, 0.2, 0.3} for RDT; Max. tree depth {5, 10, 20, 50, 100}, number of boosting rounds {10, 20, 50, 100}, learning rate {0.1, 0.2, 0.3} for XDCC, ML-XGB, XGB-BR, XGB-CC; Split methods in Table 3 for XDCC, ML-XGB. All remaining parameters were set to default values.
The Friedman test passed at \(\alpha =0.05\) and the Nemenyi critical distance is 5.18.
The Friedman test passed at \(\alpha =0.05\) for all measures but the critical distance of 1.66 is only reached for some comparisons to XDCC\(_{std}\).
The Friedman test passed for HA and SA at \(\alpha =0.05\), the critical distance is 3.05.
References
Bogatinovski, J., Todorovski, L., Dzeroski, S., Kocev, D. (2021). Comprehensive comparative study of multi-label classification methods. CoRR https://arxiv.org/abs/2102.07113
Bohlender, S., Loza Mencía, E., Kulessa, M.(2020). Extreme gradient boosted multi-label trees for dynamic classifier chains. In: Appice, A., Tsoumakas, G., Manolopoulos, Y., Matwin, S. (eds.) Proceedings of the 23rd International Conference of Discovery Science (DS-20). pp. 471–485. Springer, Thessaloniki, Greece , https://doi.org/10.1007/978-3-030-61527-7_31
Boutell, M.R., Luo, J., Shen, X., Brown, C.M.C.M. (2004). Learning multi-label scene classification. Pattern Recognition 37(9), 1757–1771 , http://www.rose-hulman.edu/~boutell/publications/boutell04PRmultilabel.pdf
Breiman, L. (2001). Random forests. Machine Learning, 45(1), 5–32.
Chen, T., Guestrin, C .(2016). XGBoost: A scalable tree boosting system. In: Proc. of the 22nd SIGKDD Int. Conf. on Knowledge Discovery and Data Mining. pp. 785–794. ACM
da Silva, P.N., Gonçalves, E.C., Plastino, A., Freitas, A.A.(2014). Distinct chains for different instances: An effective strategy for multi-label classifier chains. In: Proceedings of the European Conference on Machine Learning and Principles and Practice of Knowledge Discovery in Databases (ECML/PKDD). pp. 453–468. Springer
Dembczyński, K., Cheng, W., Hüllermeier, E.(2010). Bayes optimal multilabel classification via probabilistic classifier chains. In: Proceedings of the 27th International Conference on International Conference on Machine Learning (ICML). pp. 279–286
Dembczyński, K., Waegeman, W., Cheng, W., & Hüllermeier, E. (2012). On label dependence and loss minimization in multi-label classification. Machine Learning, 88(1–2), 5–45.
Fan, W., Greengrass, E., McCloskey, J., Yu, P.S., Drammey, K.(2005). Effective estimation of posterior probabilities: Explaining the accuracy of randomized decision tree approaches. In: Proceedings of the 5th IEEE International Conference on Data Mining (ICDM). pp. 154–161
Fan, W., Wang, H., Yu, P.S., Ma, S.(2003). Is random model better? On its accuracy and efficiency. In: Proceedings of the 3rd IEEE International Conference on Data Mining (ICDM). pp. 51–58
Friedman, J. H. (2002). Stochastic gradient boosting. Computational Statistics & Data Analysis, 38(4), 367–378.
Fürnkranz, J. (1999). Separate-and-conquer rule learning. Artificial Intelligence Review, 13(1), 3–54.
Godbole, S., Sarawagi, S.(2004). Discriminative methods for multi-labeled classification. In: Advances in Knowledge Discovery and Data Mining, 8th Pacific-Asia Conference, PAKDD 2004, Sydney, Australia, May 26-28, 2004, Proceedings. pp. 22–30
Goncalves, E.C., Plastino, A., Freitas, A.A.(2013). A Genetic Algorithm for Optimizing the Label Ordering in Multi-label Classifier Chains. In: Proceedings of the IEEE 25th International Conference on Tools with Artificial Intelligence. pp. 469–476
Joachims, T.(1998). Text categorization with suport vector machines: Learning with many relevant features. In: Machine Learning: ECML-98, 10th European Conference on Machine Learning (LNCS 1398). pp. 137–142. Springer , hdl.handle.net/2003/2595
Kong, X., Yu, P.S. (2011). An Ensemble-based Approach to Fast Classification of Multi-label Data Streams. In: Proceedings of the 7th International Conference on Collaborative Computing: Networking, Applications and Worksharing. pp. 95–104 (October)
Kulessa, M., Loza Mencía, E.(2018). Dynamic classifier chain with random decision trees. In: Proceedings of the 21st International Conference of Discovery Science (DS-18)
Kumar, A., Vembu, S., Menon, A. K., & Elkan, C. (2013). Beam search algorithms for multilabel learning. Machine Learning, 92(1), 65–89.
Li, N., Zhou, Z. (2013). Selective Ensemble of Classifier Chains. In: Multiple Classifier Systems: 11th International Workshop on Multiple Classifier Systems, pp. 146–156
Liu, W., & Tsang, I. (2015). On the optimality of classifier chain for multi-label classification. Advances in Neural Information Processing Systems, 28, 712–720.
Llerena, J.V., Deratani Mauá, D.(2017). On using sum-product networks for multi-label classification. In: Proc. of the Brazilian Conference on Intelligent Systems (BRACIS). pp. 25–30
Loza Mencía, E., & Janssen, F. (2016). Learning rules for multi-label classification: a stacking and a separate-and-conquer approach. Machine Learning, 105(1), 77–126.
Loza Mencía, E., Park, S. H., & Fürnkranz, J. (2010). Efficient voting prediction for pairwise multilabel classification. Neurocomputing, 73(7–9), 1164–1176.
Madjarov, G., Kocev, D., Gjorgjevikj, D., & Džeroski, S. (2012). An extensive experimental comparison of methods for multi-label learning. Pattern Recognition, 45(9), 3084–3104.
Malerba, D., Semeraro, G., Esposito, F.(1997). A multistrategy approach to learning multiple dependent concepts. In: Machine Learning and Statistics: The Interface, chap. 4, pp. 87–106
Mena, D., Montañés, E., Quevedo, J.R., Coz, J.J.d.(2015). Using A* for inference in probabilistic classifier chains. In: Proceedings of the 24th International Conference on Artificial Intelligence. pp. 3707–3713
Mena, D., Montañés, E., Quevedo, J.R., Coz, J.J.d.(2016). An overview of inference methods in probabilistic classifier chains for multilabel classification. Wiley Interdisciplinary Reviews: Data Mining and Knowledge Discovery 6(6), 215–230
Moyano, J.M., Gibaja, E.L., Ventura, S.(2017). MLDA: A tool for analyzing multi-label datasets. Knowledge-Based Systems 121, 1–3 , https://github.com/i02momuj/MLDA
Nam, J., Kim, J., Loza Mencía, E., Gurevych, I., Fürnkranz, J.(2014). Large-scale multi-label text classification - revisiting neural networks. In: Proceedings of the European Conference on Machine Learning and Principles and Practice of Knowledge Discovery in Databases (ECML/PKDD). pp. 437–452
Nam, J., Kim, Y., Loza Mencía, E., Park, S., Sarikaya, R., Fürnkranz, J.(2019). Learning context-dependent label permutations for multi-label classification. In: Proceedings of the 36th International Conference on Machine Learning (ICML-19). pp. 4733–4742
Nam, J., Loza Mencía, E., Kim, H.J., Fürnkranz, J.(2017). Maximizing subset accuracy with recurrent neural networks in multi-label classification. In: Advances in Neural Information Processing Systems 30 (NIPS-17). pp. 5419–5429
Nguyen, V.L., Hüllermeier, E., Rapp, M., Loza Mencía, E., Fürnkranz, J.(2020). On aggregation in ensembles of multilabel classifiers. In: Appice, A., Tsoumakas, G., Manolopoulos, Y., Matwin, S. (eds.) Proceedings of the 23rd International Conference on Discovery Science. pp. 533–547. Springer, Cham (Oct)
Papagiannopoulou, C., Tsoumakas, G., Tsamardinos, I.(2015). Discovering and exploiting deterministic label relationships in multi-label learning. In: Proc. of the 21th ACM SIGKDD Int. Conf. on Knowledge Discovery and Data Mining. pp. 915–924
Quevedo, J. R., Luaces, O., & Bahamonde, A. (2012). Multilabel classifiers with a probabilistic thresholding strategy. Pattern Recognition, 45(2), 876–883.
Rapp, M., Loza Mencía, E., Fürnkranz, J., Nguyen, V.L., Hüllermeier, E.(2020). Learning gradient boosted multi-label classification rules. In: Proceedings of the European Conference on Machine Learning and Principles and Practice of Knowledge Discovery in Databases (ECML/PKDD)
Read, J., Martino, L., & Luengo, D. (2014). Efficient Monte Carlo methods for multi-dimensional learning with classifier chains. Pattern Recognition, 47(3), 1535–1546.
Read, J., Pfahringer, B., Holmes, G., & Frank, E. (2011). Classifier chains for multi-label classification. Machine Learning, 85(3), 333–359.
Read, J., Pfahringer, B., Holmes, G., & Frank, E. (2021). Classifier chains: A review and perspectives. Journal of Artificial Intelligence Research, 70, 683–718.
Schapire, R. E., & Singer, Y. (2000). Boostexter: A boosting-based system for text categorization. Machine Learning, 39(2/3), 135–168.
Senge, R., Del Coz, J.J., Hüllermeier, E.(2014). On the problem of error propagation in classifier chains for multi-label classification. In: Spiliopoulou, M., Schmidt-Thieme, L., Janning, R. (eds.) Data Analysis, Machine Learning and Knowledge Discovery, pp. 163–170
Si, S., Zhang, H., Keerthi, S.S., Mahajan, D., Dhillon, I.S., Hsieh, C.J. (2017). Gradient boosted decision trees for high dimensional sparse output. In: Proceedings of the 34th International Conference on Machine Learning (ICML). pp. 3182–3190. PMLR
Sucar, L. E., Bielza, C., Morales, E. F., Hernandez-Leal, P., Zaragoza, J. H., & Larrañaga, P. (2014). Multi-label classification with Bayesian network-based chain classifiers. Pattern Recognition Letters, 41, 14–22.
Trajdos, P., Kurzynski, M.(2019). Dynamic classifier chains for multi-label learning. In: Fink, G.A., Frintrop, S., Jiang, X. (eds.) Proceedings of the 41st DAGM German Conference on Pattern Recognition (GCPR). pp. 567–580. Springer
Tsoumakas, G., Katakis, I., Vlahavas, I.(2010). Mining Multi-label Data. In: Data Mining and Knowledge Discovery Handbook, pp. 667–685
Tsoumakas, G., & Katakis, I. (2007). Multi-label classification: An overview. International Journal of Data Warehousing and Mining, 3(3), 1–17.
Vens, C., Struyf, J., Schietgat, L., Džeroski, S., & Blockeel, H. (2008). Decision trees for hierarchical multi-label classification. Machine Learning, 73(2), 185.
Waegeman, W., Dembczyński, K., & Hüllermeier, E. (2019). Multi-target prediction: a unifying view on problems and methods. Data Mining and Knowledge Discovery, 33(2), 293–324.
Zhang, M., & Zhou, Z. (2014). A review on multi-label learning algorithms. IEEE Transactions on Knowledge and Data Engineering, 26(8), 1819–1837.
Zhang, X., Fan, W., Du, N.(2015). Random decision hashing for massive data learning. In: Proceedings of the 4th International Workshop on Big Data, Streams and Heterogeneous Source Mining: Algorithms, Systems, Programming Models and Applications. pp. 65–80
Zhang, X., Yuan, Q., Zhao, S., Fan, W., Zheng, W., Wang, Z.(2010). Multi-label classification without the multi-label cost. In: Proceedings of the SIAM International Conference on Data Mining (SDM). pp. 778–789
Zhang, Z., Jung, C.(2019). GBDT-MO: Gradient Boosted Decision Trees for Multiple Outputs. http://arxiv.org/abs/1909.04373
Funding
Open access funding provided by Johannes Kepler University Linz. The authors declare that they have no funding.
Author information
Authors and Affiliations
Contributions
Eneldo Loza Mencía conceived the basic idea behind the proposed algorithms and led the implementation, the experimental evaluation, and the write-up. Moritz Kulessa and Simon Bohlender implemented algorithms and conducted the experiments. All authors contributed to the interpretation of the results and the write-up of the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Editors: Annalisa Appice, Grigorios Tsoumakas.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A. Appendix
A. Appendix
The tables and figures in the appendix extend the results shown previously for more datasets or for more algorithms. Fig. 11 shows the influence of the activated label tests in the RDTs on all available datasets (cf. Sect. 4.4.2). Tables 7, 8 and 9 show the performances for all algorithms on all datasets. ML-RDT denotes the RDT variant for multi-label classification introduced in Sect. 4.1, RDT-LP predicts label sets in the leafs (label powerset transformation).

Graphs for all datasets for the experiment described in Sect. 4.4.2
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Loza Mencía, E., Kulessa, M., Bohlender, S. et al. Tree-based dynamic classifier chains. Mach Learn 112, 4129–4165 (2023). https://doi.org/10.1007/s10994-022-06162-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10994-022-06162-3