
Abstract
Crimean-Congo hemorrhagic fever (CCHF) is a widespread zoonotic viral disease, caused by a tick-born virus Crimean-Congo hemorrhagic fever virus (CCHFV). This disease is endemic in Middle East, Asia, Africa and South-Eastern Europe with the mortality rate of 5–30%. CCHFV genome is composed of three segments: large, medium and small segments. M segment encodes a polyprotein (glycoprotein) so called glycoprotein N (Gn) which is considered as a potential druggable target for the effective therapy of CCHF. The complete structure of Gn is still not characterized. The aim of the current study is to predict the complete three-dimensional (3D-) structure of CCHFV Gn protein via threading-based modeling and investigate the residues crucial for binding with CCHFV envelop. The developed model displayed excellent stereo-chemical and geometrical properties. Subsequently structure based virtual screening (SBVS) was applied to discover novel inhibitors of Gn protein. A library of > 1300 anti-virals was selected from PubChem database and directed to the predicted binding site of Gn. The SBVS results led to the identification of thirty-seven compounds that inhibit the protein in computational analysis. Those 37 hits were subject to pharmacokinetic profiling which demonstrated that 30/37 compound possess safer pharmacokinetic properties. Thus, by specifically targeting Gn, less toxic and more potent inhibitors of CCHFV were identified in silico.
Similar content being viewed by others

Introduction
Crimean-Congo hemorrhagic fever (CCHF) is a prevalent and zoonotic viral disease, caused by Crimean-Congo hemorrhagic fever virus (CCHFV) which is a tick-borne virus belongs to the Nairovirus genus of Bunyaviridae family. CCHF is widespread over a range of geographical area including Middle East, Asia, Africa and South-Eastern Europe with the mortality rate of 40% (Appannanavar and Mishra 2011; Zivcec et al. 2016). After Dengue, CCHFV is the second most prevalent arbovirus with significant medical importance (Ergonul 2006; Ergonul 2012; Bente et al. 2013). The CCHFV causes severe viral haemorrhagic fever outbreaks, with a case fatality rate of 10–40% (WHO reports 2013). In Pakistan, since 2000, 50–60 cases are reported annually (WHO reports 2013; Begum et al. 1970). CCHFV spread through Hyalomma tick, a vector responsible for viral transmission. CCHFV do not show symptoms in animals, while cause mild to highly fatal disease in humans (Bente et al. 2013). The infection is usually initiated by the skin lesions produced by the infected tick. After short-term incubation period (usually ≤ 7 days) non-specific symptoms of CCHFV are initiated, including high fever, faintness, chills, irritability, limb, head and spinal pains, which can last for 5–12 days. Moreover diarrhoea, vomiting, abdominal pain, thrombocytopenia, bradycardia and elevation of circulating enzymes of liver are some other recurrent side effects of CCHF. The hemorrhages like ecchymosis, epistaxis, cerebral, gingival, and gastrointestinal haemorrhages usually begin on the fourth day of infection. The liver turns to be swollen and painful. In current infections, aspiratory hemorrhages, neurological entanglements and blood loss leads to death. Distribution of intravascular coagulopathy, multi-organ failure and shock typically results in the fatal outcome (Burt et al. 1997; Saijo et al. 2010; Bente et al. 2013). Moreover, endothelial cells, mononuclear phagocytes and hepatocytes are the main cellular targets of infection. Ribavirin is the only available antiviral drug used for the treatment of CCHF which inhibit CCHFV replication cycle, however may cause neurological and hematological anomalies (Ergonul et al. 2006; Ergonul 2008). The viral load is prominent in blood during the initial stage of CCHF, thus it is expected that the transfer of antibodies against CCHFV can be an effective therapy (Wölfel et al. 2007), however use of monoclonal antibodies against CCHFV is still in its infancy.
CCHFV is pleomorphic virus (~ 90–100 nm in diameter), consists of tripartite single-stranded negative-sense RNA genome (vRNA) which is composed of small (S), medium (M) and large (L) segments (Guo et al. 2012; Shtanko et al. 2014; Goedhals et al. 2015). This virion contains cell-membrane derived envelope (coated by mature glycoproteins) which contains genomic ribonucleoprotein complexes (RNPs) formed by vRNA, nucleoprotein and RdRp (Zivcec et al. 2016). The viral capsid is enveloped by 5 nm thick lipid bilayer and small projections (~ 5–10 nm long) are formed by the envelope proteins (Sanchez et al. 2002; Bergeron et al. 2007). The L segment is 11–14.4 kilobases in length which encodes RNA-dependent RNA polymerase (RdRp) and the nucleoprotein (NP). RdRp is required for mRNA and cRNA synthesis, necessary for translation and genome replication, respectively (Honig et al. 2004). The M segment is 4.4–6.3Kbs long which encodes envelope glycoproteins precursor (Zivcec et al. 2016) which is cleaved co-translationally by signal peptidase and then post-translationally modified into two structural trans-membrane glycoproteins (Gc and Gn), non-structural M protein (NSM), and secreted non-structural proteins (GP160, GP85 and GP38). These structural proteins form complexes on the surface of virion and assist in attachment to the host cell surface receptors, thus leads to fusion of viral envelope with the host membrane (Sanchez et al. 2002; Bergeron et al. 2007). The S segment is 1.7–2.1 kb in size, and encodes nucleocapsid protein which serves in the encapsidation of viral RNA (vRNA) and complimentary RNA (cRNA) during transcription and replication of genome. The mutation rates for the three parts of the genome were estimated to be: 1.09 × 10− 4, 1.52 × 10− 4 and 0.58 × 10− 4 substitutions/site/year for the S, M, and L segments, respectively (Carter et al. 2012).
The M segment is a crucial part of CCHFV because its Gn and Gc proteins aids CCHFV entry and fusion, formation of virion particle and immune evasion (Bertolotti-Ciarlet et al. 2005). Gn binds with ribonucleoproteins in vitro through its cytoplasmic tail that contains a zinc finger domain, implying that Gn may involve in genome packaging and has important role in viral assembly (Erickson et al. 2007). The Gn glycoprotein contains a 176 residue ectodomain followed by a 24 residues transmembrane region and terminates in a long cytoplasmic tail consisting of ~ 100 residues (Estrada and Guzman 2011; Strandin et al. 2013). Because of significant importance of Gn segment, designing a small molecule inhibitor against Gn segment could be a better approach to inhibit CCHFV. Due to lack of complete structure of Gn segment, we carried out its structural elucidation by in silico homology modeling. The generated model was used to identify novel drug like compounds against CCHFV by targeting Gn protein. Subsequently we performed ADMET profiling of the selected hits. This study has resulted in the discovery of novel scaffolds against CCHFV Gn protein.
Materials and Methods
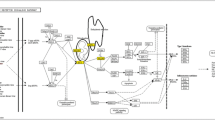
Threading based modeling was conducted by I-TASSER (Yang et al. 2015), RaptorX (Wang et al. 2016a) and ModBase (Pieper et al. 2010) servers. Molecular Operating Environment (MOEv2014.09) was used for Molecular docking. PLIF (Protein-Ligand Interaction Fingerprints) (Labute 2001) utility was used for protein ligand interaction calculation. The illustrations of 3D- model and Protein-ligand interactions were prepared by UCSF Chimera (Pettersen et al. 2004). The complete strategy is depicted in Fig. 1.

Schematic presentation of computational work flow
Sequence Retrieval of CCHFV Gn Protein
M-segment of CCHFV codes for polyprotein glycoprotein. The sequence of M segment was retrieved from NCBI with the accession code ARB51455. This sequence consists of 1684 residues; Gn sequence is resided between 520 and 842 region, which was selected from UniProtKB (code: Q8JSZ3) for modeling. The Gn sequence is given below:
F > sp|Q8JSZ3|GP_CCHFI Envelopment polyprotein OS = Crimean-Congo hemorrhagic fever virus (strain Nigeria/IbAr10200/1970) GN = GP PE = 3 SV = 1.
SEEPSDDCISRTQLLRTETAEIHGDNYGGPGDKITICNGSTIVDQRLGSELGCYTINRVRSFKLCENSATGKNCEIDSVPVKCRQGYCLRITQEGRGHVKLSRGSEVVLDACDTSCEIMIPKGTGDILVDCSGGQQHFLKDNLIDLGCPKIPLLGKMAIYICRMSNHPKTTMAFLFWFSFGYVITCILCKAIFYLLIIVGTLGKRLKQYRELKPQTCTICETTPVNAIDAEMHDLNCSYNICPYCASRLTSDGLARHVIQCPKRKEKVEETELYLNLERIPWVVRKLLQVSESTGVALKRSSWLIVLLVLFTVSLSPVQSAPI
Three Dimensional (3D)-Structure Prediction
For homology modeling, template searching was carried out on BLAST protein (with psi blast option) which retrieved sequences of very low coverage i.e., < 30%. Therefore modeling of Gn segment was performed by threading based approach. For this purpose, I-TASSER server was used in which proteins models are developed based on the structural alignment of the selected templates, instead of sequence alignment and homology (percent identity). The best model (obtained by I-TASSER) was validated by Procheck Ramachandran plot to predict the stereochemical properties of the protein. The results were not accurate thus RaptorX server was used to predict the reliable structure. RaptorX use multiple templates threading protocol to build 3D model from single target sequence and the quality of the final model is improved by its ability to correct the errors partially in pairwise alignments and alignment coverage is also increased. The RaptorX generated model was again evaluated by Procheck, which showed good results. However RaptorX generated model possess several disordered loops, therefore, two loops starting from residues 38–96 and 274–323 were separately modeled. After several attempts on these three servers, we obtained best model of region 38–96 and 274–323 from ModBase and RaptorX, respectively. The structural properties of the predicted loops were evaluated and replaced in the 3D-model of Gn by Chimera. The binding site of Gn segment was predicted by COACH server (Yang et al. 2013) and by aligning the binding site with the 2L7X.
Compound Selection for Virtual Screening
A set of 1392 anti-viral compounds were retrieved from PubChem database for virtual screening. The chemical structures of compounds were checked by MOE and converted into 3D by MOE wash module, hydrogen atoms were added and partial charges were assigned on each structure. The compounds were minimized to their lowest energy conformation by using MMFF94x force field until the gradient was reached to 0.01 RMS/ kcal/mol.
Molecular Docking
The selected library of compounds was docked in the predicted Gn binding site by MOE. Using Protonate3D module, ionization state of model was assigned, hydrogens were added and electrostatic potential was calculated. Subsequently model was minimized using MMFF94x force field with the default parameters. For docking, virtual screening protocol of MOE was used with Triangle Matcher placement method, London dG scoring function and force field (GBVI/WSA dG) refinement method. Finally protein-ligand interactions were calculated by PLIF. The interactions were rendered by Chimera.
Results
Threading Based Modeling
The 3D-structure of Gn protein was initially elucidated through I-TASSER, however the stereochemical properties of the obtained model was not good, thus we use RaptorX server for modeling. RaptorX built secondary and tertiary structures of query sequence by aligning it with the sequences of multiple templates and the quality of the predicted model is evaluated by p-value, Score, uGDT and GDT (Peng and Xu 2011; Ma et al. 2013). The 323 amino acid sequence of Gn glycoprotein of CCHFV was submitted to RaptorX to model the structure of Gn. The Gn model retrieved from RaptorX contains three domains, each domain was deduced by aligning one or more top matched template sequences present in the template library. The model is composed of an ectodomain (residue Ser1-Pro168), a transmembrane domain (residues Lys169-Leu206), a zinc finger cytoplasmic domain (residues Lys207-Glu278), and a cytoplasmic tail (residues Arg279-Ile323). The templates used to construct the model are 2L7X (Chain A) (Estrada and Guzman 2011), 5M87 (Chain A) (Ehrnstorfer et al. 2017), 2A65 (Chain A) (Yamashita et al. 2005) and 5G47 (Chain A) (Halldorsson et al. 2016). Alignment of the templates and the query sequence is depicted in Fig. 2. Properties of the templates are shown in Table 1. GDT and uGDT are the global distance test and unnormalized global distance test, respectively. The absolute model quality is measured by uGDT (GDT). If uGDT is > 50 then it is a good indicator for the protein having residues more than 100. GDT > 50 is a good indicator for the protein having residues < 100. Thus, if a good uGDT (> 50) is shown by a model but at the same time it has bad GDT (< 50) then it means that only a small portion of the protein model is good. The score indicate the alignment score that range from 0 to the domain sequence length while “0” score shows the results to be worst. Relative quality of the model is predicted by the p-value that should be < 10− 3 and 10− 4 for mainly α and β proteins, respectively. The majority of the domains consist of α helix and none of the domains has p-value > 10− 3 that shows the quality of model is good. The predicted structure of model is shown in Fig. 3. The structure of the model is composed of three domains and the best template was 5G47 (chain A). Overall uGDT (GDT) value of the predicted structure is 126 (39) in which the GDT value is 39 which is < 50 and uGDT value is 126 i.e., > 50. This indicates that the small portion of the whole model is good. The complete sequence was (100% residues) modeled by RaptorX, out of which only 17% residues were predicted as disorder, suggesting that the rest 87% residues are in ordered position. The ordered and disordered positions are depicted as blue and red bars in Fig. 4.

The sequence alignment of CCHFV Gn Protein and templates

The 3D-structure of Gn protein predicted by (a) iTasser (b) Raptor X (c) Gn38-96 predicted by Modbase (d) Gn274-323 predicted by RaptorX (e) Final 3D-structure of Gn Protein (f) The superimposed view of 2L7X template (blue) and Gn model (Gold) and the conserved binding residues are highlighted in circle (Color figure online)

The quality of each residue of model. Ordered and disordered positions in the model are shown in blue and maroon color, respectively (Color figure online)
Secondary Structure Prediction
The secondary structure of the Gn model was deduced by RaptorX (Wang et al. 2016b; Schaarschmidt et al. 2017) which display result in two modes: (i) 3-state secondary structure and (ii) 8-state secondary structure. The 3-state secondary structure is comprised of α-helix, β-sheet and coiled regions represented by H, E and C respectively. The secondary structure of Gn shows that it contains 37% H, 24% E and 37% C (Fig. 5).

The predicted secondary structure by using 3-class secondary structure method. The helix, β-sheets and coils are presented as red, blue and grey bars, respectively (Color figure online)
Solvent Accessibility of the Model
The solvent exposed areas of the model was predicted by 3-state solvent accessibility method of RaptorX (Yang et al. 2018), which shows solvent exposed, medium and buried residues as E, M and B, respectively. The solvent accessibility of Gn shows that 31% region was exposed, 48% was resided in medium region while 19% region was laid in buried region (Fig. 6).

The predicted solvent accessibility of the model. Blue, Yellow and Red color shows buried, medium and exposed portions, respectively (Color figure online)
Model Validation and Loops Modeling
The stereo-chemical properties of the Gn model was validated by PROCHECK-Ramachandran plot, which shows that 86.6% of total residues were present in most favored region and 11.3% residues were in additional allowed regions and 1.4% residues were in generously allowed regions while only 0.7% residues were present in disallowed regions. The Ramachandran plot is shown in supporting information Fig. S1. The 3D-model predicted by RaptorX was composed of several loops (Fig. 3) hence two loops were modeled separately by different programs. Therefore, regions that contain > 50 residues were modeled again. Thus Gn (38–96) region was modeled by Modbase server using 5B0U (Chain A) as template with 53% identity with region 38–96. The model (38–96) showed that this loop is not composed of coils, but three anti-parallel β-sheets. The model quality was assessed by Ramachandran plot (Fig. S2) shows that 92% residues were present in most favored region while none of the residues were in disallowed region. The region Gn (274–323) was also modelled by RaptorX using 5UAK (Chain A), 5UAR (Chain A) and 4WAT (Chain A) as templates, and the resultant model shows that this region (274–323) is composed of 68% helical part. The Ramachandran plot (Fig. S3) shows that 97.8% residues lie in the most favored region while only 1(2.2%) residue is present in the disallowed region. These modeled segments were then replaced and joined into the initial model of Gn (predicted by RaptorX) by using Chimera. The complete structure after loops modeling is shown in Fig. 3. The final model of the Gn was then validated by Ramachandran plot, depicted that 88.4% residues lie in the most favored region, while 9.2%, 1.4%, and 1.1% residues were present in additional allowed, generously allowed and disallowed region respectively. The stereo-chemical properties indicate that the model is of good quality (Fig. S4).
Prediction of the Binding Site of the Model
The residues involve in the protein-protein, or protein-ligand binding were identified by using COACH algorithm (Wu et al. 2018) which uses structural alignment method to predicts the binding site of the protein by comparing the binding sites of different templates. These sites are predicted by two methods namely: TM-SITE and S-SITE. According to COACH results, binding site are located on region 217, 220, 233, and 237 of Gn glycoprotein and its corresponding template is 2L7X (Chain A). The template 2L7X:A contains two CCHC-type zinc finger domains, among them the first zinc finger corresponding residues are Cys36, Cys39, His52, Cys56 and the second zinc finger corresponding residues are Cys61, Cys64, His76 and Cys80 [59]. The alignment of the active site of 2L7X and Gn-model depicted that Cys36, Cys39, His52 and Cys56 of 2L7XA aligned with Cys217, Cys220, His233 and Cys237 of Gn model, while Cys61, Cys64, His76 and Cys80 of 2L7XA aligned with Cys242, Cys245, His257 and Cys261 of Gn model. The results are tabulated in Table 2.
Virtual Screening
A set of 1392 compounds with anti-viral activites were retrieved from PubChem and screened against Gn protein by molecular docking. Based on docking rank and score, top 500 compounds were selected for their interactions analysis. The PLIF calculated results showed that thirty seven compounds interact precisely with the zinc finger domain of Gn protein. The docking and PLIF results of the selected hits are tabulated in Table 3.
ADMET Prediction of Selected Hits
The pharmacokinetic properties of selected hits were evaluated by ADMETsar (http://lmmd.ecust.edu.cn/admetsar1/predict/) and SwissADME (http://www.swissadme.ch/). The results are tabulated in Table 4. The results depicted that only six compounds (23, 28–32 and 36) displayed AMES toxicity while the rest are non-carcinogenic. Moreover five compounds (29–32 and 36) are predicted to cross blood brain barrier (BBB) while the rest do not show BBB positivity. Among thirty seven hits, twelve compounds (22–25, 28–34 and 36) displayed high intestinal absorption in humans. The predicted acute toxicity in rat models showed that the compounds do not show lethality upto the concentration of 2 mol/Kg, hence we can say that these compounds are not lethal in lower doses and fall in the good range of median lethal dose (LD50). The ADMETsar server predicted the acute oral toxicity of all the compounds. According to the results, compound 31 fall in category II (50 mg/kg > LD50 < 500 mg/kg) and compound 13 and 19 fall in category IV (LD50 = ≥ 5000 mg/kg). However the rest of the compounds fall in class III (LD50 = > 500 mg/kg, ≤ 5000 mg/kg). The results indicate that the compounds do not show oral toxicity on doses up to 5000 mg/kg, thus the compounds are not orally toxic. The predicted metabolic profile of the compounds shows that which cytochrome p450 will act as substrate and non-substrate for the compound and which will be inhibited by the compound. The molecules with high AMES toxicity and high BBB permeability were excluded from selection. The final selected compounds with their respective ADMET properties are tabulated in Table 4.
Interaction Analysis of the Selected Hits
Followed by the interaction analysis, compounds were segregated into two categories. The compounds which bind with the binding site were categorized as category I, while compounds that particularly interact with the residues of zinc finger domain were classified as category II. The compounds 1, 2, 4–6, 9–11, 13, 18–21, 22, 25–27, and 35 are included in category I. The binding mode of compound 1 showed that compound binds with Arg219 and Arg211 through H-bond and ionic bond, respectively. The compound 2 interacts with Cys224 and Lys226 via H-bond and ionic bond, respectively, while compound 4 accepts H-bonds from Gln223 and Lys226, and mediates H-bond with Cys224 and Asp215. The compound 5 formed H-bond with Cys224 and Arg219. Compound 6 accepts H-bond from the side chain of Arg211 and Ser210, and formed ionic bonding with Lys226. The side chain of His220 provides hydrophobic interactions to the compound to stabilize it in the binding region. Compound 9 accepts H-bond from Lys226, Gln223 and Arg211. Moreover compound is stabilized in the binding site by hydrophobic interaction provided by side chain of His220. The compound 10 formed H-bond with Cys224, His 220 and Arg211, and an ionic interaction was observed between Arg211 and the ligand. Compound 11 mediates H-bonding with Ser210, Cys208 and Arg219, an ionic interaction with Lys226 and a hydrophobic interaction with His220. The compound 13 interacts with Arg219 and Lys226 through H-bonding and hydrophobically by His220. Compound 18 mediates H-bonding with Arg211, Ser210 and His220, while compound 19 formed ionic bond with Arg211and Lys226, and H-bond with Cys224. Compound 20 interacts with Lys226 and Cys224 via H-bond, while compound 21 interacts with Lys226 through ionic bond and H-bonding. The compound 22 formed H-bond with Lys226 and Cys224, and a hydrophobic interaction with the side chain of Tyr207. The compounds 25, 26, 27 and 35 depicts that these compounds formed ionic bond with Lys226 and Arg211, while compound 27 also formed arene-hydrogen interaction with Ser210. The binding interactions of these compounds are shown in supporting information Fig. S5A-S5R.
Compound 3, 7–8, 12, 14–17, 24, 33–34, and 37 are included in category II, which interact with zinc finger domain of Gn-protein (Fig. 7). The docked view of compound 3 depicts that the compound mediates H-bonds with Tyr207 (2.5 Å) and Ala209 (2.7 Å), and an ionic bond with Cys208 (2.1 Å). Moreover the benzoyl-OH moiety of the compound mediates bi-dentate interactions with Arg211 (1.5 Å and 2.5 Å). The docked view of compound 7 showed that weak H-bond is formed between carbonyl oxygen and Cys208 (3.3 Å) and a Π–Π interaction is formed between the phenyl moiety of the ligand and His220 (2.1 Å). The phenyl-OH of the compound is H-bonded with Arg211 (0.7 Å). The collective H-bonds and hydrophobic interactions are responsible for the inhibition of zinc finger domain. The binding mode of compound 8 depicts that compound mediates weak H-bond with Glu229 (3.1 Å) and a strong H-bond between carbonyl oxygen and the side chain of His220 (1.6 Å). Additionally several residues of zinc finger domain provides hydrophobic interactions to stabilize the compound. The compound 12 mediates several H-bonds within zinc finger domain. The sulfate moiety of compound forms bi-dentate interactions with Ser210 (1.8 Å and 2.5 Å), and two H-bonds with Cys208 (2.8 Å) and His220 (2.4 Å). Moreover ligand formed bi-dentate interactions with Glu223 at a distance of 1.6 Å and 1.9 Å, respectively. The carbonyl oxygen of the ligand formed a strong H-bond with Arg219 (1.7 Å). The predicted binding mode suggests that this compound could be a potent inhibitor of CCHFV-Gn protein because of these multiple interactions. The carbonyl oxygen at 3-methyl-butanoyl moiety of compound 14 mediates H-bond with His220 (2.3 Å) and Cys208 (2.4 Å). Additionally compound also forms Π–Π interactions with the side chain of Tyr207. The 4-fluoro-3-hydroxyne-4-methyloxolan and 2, 4-dioxopyrimidin moieties of compound 15 accepts H-bond from the side chain of His220 (2.3 Å) and Cys208 (2.4 Å), respectively. The trihydroxybenzoyl moiety of the compound 16 mediates ionic interaction with Lys226 (2.3 Å), and the carbonyl oxygen of the compound formed H-bond with the –OH of Ser210 (1.9 Å). The benzoyl moiety of compound forms H-bond with Gln223 and His220 at a distance of 2.2 Å and 2.6 Å, respectively. The compound 17 mediates multiple H-bonding with the surrounding residues. The tri-hydroxyphenyl moiety of the compound formed H-bonds with Cys208 (2.4 Å), Ser210 (2.6 Å) and His220 (1.7 Å). The tri-hydroxy phenyl substituted carbonyl moiety interacts with Gln223 (2.9 Å), thus compound 17 also form multiple strong interactions with the zinc finger domain. The compound 24 mediates strong H-bond with the side chains of Tyr207 (1.9 Å), His220 (1.9 Å) and Arg219 (2.5 Å). The compound 33 also mediates several hydrophilic interactions with the zinc finger domain. The carbonyl moiety of the compound formed H-bond with Cys208 (3.1 Å), and bi-dentate interactions with the side chain of Arg211 at a distance of 1.9 Å and 2.3 Å, respectively. Moreover the compound also formed bi-dentate interaction with the side chain of Ser210 (2.6 Å and 1.8 Å). Additionally several hydrophobic interactions stabilize the compound within the binding site. The compound 34 formed three H-bonds with Tyr207, His220 and Arg219. The compound is composed of four rings. One of tri-hydroxyphenyl moiety of compound interacts with Tyr207 via H-bond (1.9 Å) while carboxyl group formed H-bond with the side chain of His220 (1.9 Å). Another phenyl moiety accepts H-bond form the side chain of Arg219 (2.5 Å). The compound 37 formed H-bond with Cys208 and His220 at a distance of 2.4 Å and 1.9 Å, respectively. A weak H-bond was observed between phenyl group of the compound and Tyr207 (3.6 Å). The binding interactions of these compounds are shown in Fig. 7. The result suggests that these compounds bind with the zinc finger domain with strong interactions, thus capable to hinder the function of Gn protein.

The docked view of compounds 3, 7, 8, 12, 14, 15, 16, 17, 24, 33, 34, and 37 (a–l) that interacts with zinc finger domain
Discussion
CCHF is a life threatening viral disease with high mortality and morbidity rate (Rahpeyma et al. 2015). Though CCHFV belongs to Bunyaviridae family however comparing to other genera of this family, it shows some uncommon properties; for instance the length of the M-segment of CCHFV is a large precursor, comprised of 1684 amino acids and a remarkably large glycoprotein is encoded by this precursor protein. Another feature which distinguishes CCHFV from other genera is that its M-segment encoded glycoprotein precursor undergoes complex series of proteolysis before maturation while other viral glycoprotein undergoes proteolysis in a single step. Cysteine residues present in CCHFV glycoproteins indicate the complexity of its secondary structure due to presence of disulfide bonds (Bertolotti-Ciarlet et al. 2005; Altamura et al. 2007; Carter et al. 2012). Because of important role of Gn protein in viral assembly and localization, several researches have targeted this glycoprotein as a potent immunogen for vaccine development by using various expression systems (Saijo et al. 2010; Strandin et al. 2013; Buttigieg et al. 2014; Dowall et al. 2017; Wu et al. 2017).
The three-dimensional (3D) structure of any protein facilitates its functional characterization (Ul-Haq et al. 2015; Purohit et al. 2018; Khan and Ansari 2017; Khan et al. 2017).
To construct the model of Gn glycoprotein, I-TASSER server was used initially, however the retrieved model showed lower quality and out of 323 residues, 157 lied in most favored region, 104 in additionally allowed region, 16 in generously allowed region while 7 residues were present in the disallowed region. I-TASSER uses structural fragments of multiple templates to build the model as a result the generated model can have lower quality due to the presence of more torsion angles in their backbone. The complex and challenging proteins with less close homologous templates show such type of results (Ul-Haq et al. 2015). Since our target protein is complex, we tested RaptorX for its modeling. RaptorX server is usually used to construct models of those targets which have few close homologs by employing multiple template structure. The resultant model showed acceptable stereo-chemical profile however two loops were geometrically unacceptable in the model; those two loops were modeled separately and joined in the model.
The Gn of all nairoviruses has conserved cysteine and Histidine residues in their cytoplasmic tail (CTs) which are responsible for the formation of zinc finger domain. Nairoviruses possess dual CCHC type zinc fingers that form a globular domain by tightly associating with each other. The role of ZF’s domain is the regulation of DNA and viral RNA (Strandin et al. 2013). Andes virus (ANDV) and CCHFV contains dual zinc fingers with similar structure. The only difference is that ANDV does not have the ability to bind with viral RNA while CCHFV binds with viral RNA (Altamura et al. 2007). The glycoproteins Gn and Gc belongs to type I membrane integral proteins and extend viral membrane, its N-terminal domain contact with outer environment and act as ectodomain while C-terminal points toward intraviral space. Bunyaviruses are different from other single stranded anti-sense RNA viruses because they lack protein which acts as scaffold between viral envelope and RNP components (matrix protein). However, viral Gn-CTs are large enough that they can accommodate domains and performs function like matrix protein. So, they can be assumed as substitutes of viral matrix protein (Strandin et al. 2013). The thrombocytopenia syndrome virus and Rift Valley fever virus belongs to the Phleboviruses genus of the bunyaviridae family, their Gn is also type-I integral membrane protein and its N-terminal act as ectodomain while C-terminal domain is transmembrane helical portion that is inserted in viral membrane (Wu et al. 2017). The CCHFV Gn model also depicts that its C-terminal is helical.
The studies showed that Gn glycoprotein contains a 176 residue ectodomain followed by a 24 residues transmembrane region and a long cytoplasmic tail composed of > 100 residues (Estrada and Guzman 2011). The developed model is composed of an ectodomain (residue Ser1-Pro168), a transmembrane domain (residues Lys169-Leu206), a zinc finger cytoplasmic domain (residues Lys207-Glu278), and a cytoplasmic tail (residues Arg279-Ile323.) Several studies revealed that Gn of protein of virus from bunyaviridae family is involved in viral assembly. For example, alanine mutagenesis of the cytoplasmic tails of Uukuniemi virus and Bunyamwera virus affect the ability of virus-like particles to effectively incorporate ribonucleoproteins, thus intimating a role for Gn tails in genome packaging. More recently, the Gn tail of Puumala virus was shown to co-immunoprecipitate with the Puumala nucleocapsid protein. These results suggest that the CCHFV Gn tail plays an equally important role in viral assembly of genus Nairovirus (Sanchez et al. 2002; Estrada and Guzman 2011; Wu et al. 2017).
Drug designing is a complex process where computational tools help to foster this process in less time and automatic procedures. Due to high mutation rate of viral proteins, it is increasingly demanding to expedite the drug delivery against viral diseases. Computational medicinal chemistry is not only applied against human disease but also delivered several novel fungicides against plants diseases (Iftikhar et al. 2017). It is important to predict toxicity, ADME properties and potential activity of a drug like molecules prior to their biological testing in order to avoid drug failure. In the present study, these properties are identified by SwissADMET and ADMETsar. Virtual screening is computational searching of huge chemical space against targets. Previously structure based virtual screening was applied to identify the novel immunomodulators against human immune disorders (Halim et al. 2013) and several drugs like molecules were suggested against dengue virus to establish effective therapeutics (Halim et al. 2017). We believe that the predicted hits will be a valuable starting point to deliver drugs against congo virus.
Conclusions
CCHF is contagious disease; currently there is no drug or vaccine available to treat this fatal disease. This study was conducted to explore computational resources to get insights into the inhibitory mechanism of CCHFV. Glycoprotein Gn of CCHFV has been exploited as an important drug target in this study because of its role in viral envelop binding. The zinc finger domain of this protein is available however complete three dimensional structure of this protein is not available. Thus threading based in silico modeling was employed to elucidate its complete structure which was used for the development of new drugs by structure based virtual screening of antiviral compounds. The computational analysis revealed that out of > 1300 compounds, thirty seven compounds were compatible with the binding site and are anticipated to block the activity of Gn in silico. The in silico predicted ADMET profile suggests that thirty compounds has safer pharmacokinetic properties and could be exploited as potential hits. The results need in vitro and in vivo experimental validation to confirm these results.
Supporting Information
Ramachandran plot of Gn model, Ramachandran plot of Gn (38–96) region predicted by modbase, Ramachandran plot of the Gn (274–323) region predicted by RaptorX, Ramachandran plot of the 3D structure of the CCHFV Gn protein and the docked view of compounds 1–2, 4–6, 9–11, 13, 18–22, 25–27, 35 are included in supporting information.
References
Altamura LA, Bertolotti-Ciarlet A, Teigler J, Paragas J, Schmaljohn CS, Doms RW (2007) Identification of a novel C-terminal cleavage of Crimean-Congo hemorrhagic fever virus PreGN that leads to generation of an NSM protein. J Virol 81:6632–6642
Appannanavar SB, Mishra B (2011) An update on Crimean Congo hemorrhagic fever. J Glob Inf Dis 3:285–292
Begum F, Wisseman C Jr, Casals J (1970) Tick-Borne viruses of west Pakistan: IV. Viruses similar to, or identical with, Crimean Hemorrhagic fever (Congo-Semunya), WAD MEDANI and PAK ARGAS 461 Isolated from Ticks of the Changa Manga Forest, Lahore District, and of Hunza, Gilgit Agency, W. Pakistan. Am J Epidem 92:197–202
Bente DA, Forrester NL, Watts DM, Mcauley AJ, Whitehouse CA, Bray M (2013) Crimean-Congo hemorrhagic fever: history, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antivir Res 100:159–189
Bergeron É, Vincent MJ, Nichol ST (2007) Crimean-Congo hemorrhagic fever virus glycoprotein processing by the endoprotease SKI-1/S1P is critical for virus infectivity. J Virol 81:13271–13276
Bertolotti-Ciarlet A, Smith J, Strecker K, Paragas J, Altamura LA, Mcfalls JM, Frias-Stäheli N, García-Sastre A, Schmaljohn CS, Doms RW (2005) Cellular localization and antigenic characterization of Crimean-Congo hemorrhagic fever virus glycoproteins. J Virol 79:6152–6161
Burt FJ, Swanepoel R, Shieh WJ, Smith JF (1997) Immunohistochemical and in situ localization of Crimean-Congo hemorrhagic fever (CCHF) virus in human tissues and implications for CCHF pathogenesis. Arch Pathol Lab Med 121:839–846
Buttigieg KR, Dowall SD, Findlay-Wilson S, Miloszewska A, Rayner E, Hewson R, Carroll MW (2014) A novel vaccine against Crimean-Congo Haemorrhagic Fever protects 100% of animals against lethal challenge in a mouse model. PLoS ONE 9:e91516
Carter SD, Surtees R, Walter CT, Ariza A, Bergeron E, Nichol ST, Hiscox JA, Edwards TA, Barr JN (2012) Structure, function and evolution of the Crimean-Congo hemorrhagic fever virus nucleocapsid protein. J Virol VI:01555–01512
Dowall SD, Carroll MW, Hewson R (2017) Development of vaccines against Crimean-Congo haemorrhagic fever virus. Vaccine 35:6015–6023
Ehrnstorfer IA, Manatschal C, Arnold FM, Laederach J, Dutzler R (2017) Structural and mechanistic basis of proton-coupled metal ion transport in the SLC11/NRAMP family. Nat Commun 8:1–11
Ergonul O (2006) Crimean-Congo haemorrhagic fever. Lancet Inf Dis 6:203–214
Ergonul O, Celikbas A, Baykam N, Eren S, Dokuzoguz B (2006) Analysis of risk-factors among patients with Crimean‐Congo haemorrhagic fever virus infection: severity criteria revisited. Clin Microbiol Infect 12:551–554
Ergonul O (2008) Treatment of Crimean-Congo hemorrhagic fever. Antivir Res 78:125–131
Ergonul O (2012) Crimean-Congo hemorrhagic fever virus: new outbreaks, new discoveries. Curr Opin Virol 2:215–220
Erickson BR, Deyde V, Sanchez AJ, Vincent MJ, Nichol ST (2007) N-linked glycosylation of Gn (but not Gc) is important for Crimean Congo hemorrhagic fever virus glycoprotein localization and transport. Virology 361:348–355
Estrada DF, Guzman RND (2011) Structural characterization of the Crimean-Congo hemorrhagic fever virus Gn tail provides insight into virus assembly. J Biol Chem 286:21678–21686
Goedhals D, Bester PA, Paweska JT, Swanepoel R, Burt FJ (2015) Comparative analysis of the L, M, and S RNA segments of Crimean–Congo haemorrhagic fever virus isolates from southern Africa. J Med Virol 87:717–724
Guo Y, Wang W, Ji W, Deng M, Sun Y, Zhou H, Yang C, Deng F, Wang H, Hu Z (2012) Crimean–Congo hemorrhagic fever virus nucleoprotein reveals endonuclease activity in bunyaviruses. PNAS USA 109:5046–5051
Halim SA, Abdallah OM, Mesaik MA, Wadood A, Haq ZU, Kontoyianni M (2013) Identification of novel Interleukin-2 inhibitors through computational approaches. Mol Divers 17:345–355
Halim SA, Khan S, Khan A, Wadood A, Mabood F, Hussain J, Al-Harrasi A (2017) Targeting dengue virus NS-3 helicase by ligand based pharmacophore modeling and structure based virtual screening. Front Chem 5:1–16
Halldorsson S, Behrens AJ, Harlos K, Huiskonen JT, Elliott RM, Crispin M, Brennan B, Bowden TA (2016) Structure of a phleboviral envelope glycoprotein reveals a consolidated model of membrane fusion. PNAS USA 113:7154–7159
Honig JE, Osborne JC, Nichol ST (2004) Crimean–Congo hemorrhagic fever virus genome L RNA segment and encoded protein. Virology 321:29–35
Iftikhar S, Shahid AA, Halim SA, Wolters PJ, Vleeshouwers VGAA, Khan A, Al-Harrasi A, Ahmad S (2017) Discovering novel alternaria solani succinate dehydrogenase inhibitors by in Silico modeling and virtual screening strategies to combat early blight. Front Chem 5:1–17
Khan I, Ansari A (2017) Prediction of a highly deleterious mutation E17K in AKT-1 gene: an in silico approach. Biochem Biophys Rep 10:260–266
Khan I, Ansari A, Singh P, Dass FP (2017) Prediction of functionally significant single nucleotide polymorphisms in PTEN tumor suppressor gene: an in silico approach. Biotechnol Appl Biochem 64:657–666
Labute P (2001) Probabilistic Receptor Potentials. J Chem Comp Grp
Ma J, Wang S, Zhao F, Xu J (2013) Protein threading using context-specific alignment potential. Bioinformatics (Proceedings of ISMB 2013) 29:i257–i265
Moev2014.09. Chemical Computing Group ULC, Montreal, QC, Canada
Peng J, Xu J (2011) RaptorX: exploiting structure information for protein alignment by statistical inference. Proteins 79(suppl 10):161–171
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Pieper U, Webb BM, Barkan DT, Schneidman-Duhovny D, Schlessinger A, Braberg H, Yang Z, Meng EC, Pettersen EF, Huang CC, Datta RS, Sampathkumar P, Madhusudhan MS, Sjölander K, Ferrin TE, Burley SK, Sali A (2010) ModBase, a database of annotated comparative protein structure models, and associated resources. Nuc Acids Res 39:D465–D474
Purohit R, Kumar S, Hallan V (2018) Screening of potential inhibitor against coat protein of apple chlorotic leaf spot virus. Cell Biochem Biophys 76:273–278
Rahpeyma M, Fotouhi F, Makvandi M, Ghadiri A, Samarbaf-Zadeh A (2015) Crimean-Congo hemorrhagic fever virus Gn bioinformatic analysis and construction of a recombinant bacmid in order to express Gn by baculovirus expression system. Jund J Microbiol 8:e25502–e25511
Saijo M, Morikawa S, Kurane I (2010) Recent progress in the treatment of Crimean–Congo hemorrhagic fever and future perspectives. Fut Virol 5:801–809
Sanchez AJ, Vincent MJ, Nichol ST (2002) Characterization of the glycoproteins of Crimean-Congo hemorrhagic fever virus. J Virol 76:7263–7275
Schaarschmidt J, Monastyrskyy B, Kryshtafovych A, Bonvin AMJJ (2017) Assessment of contact predictions in CASP12: co-evolution and deep learning coming of age. Prot Struct Funct Bioinform 86:51–66
Shtanko O, Nikitina RA, Altuntas CZ, Chepurnov AA, Davey RA (2014) Crimean-Congo hemorrhagic fever virus entry into host cells occurs through the multivesicular body and requires ESCRT regulators. PLoS Pathog 10:e1004390
Strandin T, Hepojoki J, Vaheri A (2013) Cytoplasmic tails of bunyavirus Gn glycoproteins—could they act as matrix protein surrogates? Virology 437:73–80
Ul-Haq Z, Saeed M, Halim SA, Khan W (2015) 3D structure prediction of human β1-adrenergic receptor via threading-based homology modeling for implications in structure-based drug designing. PloS ONE 10:e0122223
Wang S, Li W, Liu S, Xu J (2016) RaptorX-Property: a web server for protein structure property prediction. Nucl Acids Res 44:W430–W435
Wang S, Peng J, Ma J, Xu J (2016) Protein secondary structure prediction using deep convolutional neural fields. Sci Rep 6:18962–18973
Who Reports (2013) (a) Guidelines for Crimean-Congo Hemorrhagic Fever (CCHF), Developed with joint collaboration of National Institute of Health (NIH), World Health Organization (WHO). Islamabad, 2013. (b) Seasonal awareness and alert letter for epidemic-prone infectious diseases in Pakistan, Ministry of National Health Services, Regulation and Coordination Government of Pakistan, National Institute of Health, Islamabad, 2016. www.nih.org.pk
Wölfel R, Paweska JT, Petersen N, Grobbelaar AA, Leman PA, Hewson R, Georges-Courbot MC, Papa A, Günther S, Drosten C (2007) Virus detection and monitoring of viral load in Crimean-Congo hemorrhagic fever virus patients. Emerg Infect Dis 13:1097–1100
Wu Y, Zhu Y, Gao F, Jiao Y, Oladejo BO, Chai Y, Bi Y, Lu S, Dong M, Zhang C (2017) Structures of phlebovirus glycoprotein Gn and identification of a neutralizing antibody epitope. PNAS USA 114:E7564–E7573
Wu Q, Peng Z, Zhang Y, Yang J (2018) COACH-D: improved protein–ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nuc Acids Res 46:W438–W442
Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E (2005) Crystal structure of a bacterial homologue of Na+/Cl–dependent neurotransmitter transporters. Nature 437:215–223
Yang J, Roy A, Zhang Y (2013) Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 29:2588–2595
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I-TASSER suite: protein structure and function prediction. Nat Met 12:7–8
Yang Y, Gao J, Wang J, Heffernan R, Hanson J, Paliwal K, Zhou Y (2018) Sixty-five years of the long march in protein secondary structure prediction: the final stretch? Brief Bioinf 19:482–494
Zivcec M, Scholte FE, Spiropoulou CF, Spengler JR, Bergeron É (2016) Molecular insights into Crimean-Congo hemorrhagic fever virus. Viruses 8:106–127
Author information
Authors and Affiliations
Contributions
SAH outline the research strategy and idea, drafted and revised the manuscript. SA carried out literature search, and performed computational experiments. AW provided MOE software for virtual screening. MI, AJ and AH suggested valuable comments on manuscript writing. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Halim, S.A., Aziz, S., Ilyas, M. et al. In Silico Modeling of Crimean Congo Hemorrhagic Fever Virus Glycoprotein-N and Screening of Anti Viral Hits by Virtual Screening. Int J Pept Res Ther 26, 2675–2688 (2020). https://doi.org/10.1007/s10989-020-10055-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-020-10055-1