
Abstract
Protein phosphatase (PP) type 2A is a multifunctional serine/threonine phosphatase that is involved in cardiac excitation–contraction coupling. The PP2A core enzyme is a dimer, consisting of a catalytic C and a scaffolding A subunit, which is targeted to several cardiac proteins by a regulatory B subunit. At present, it is controversial whether PP2A and its subunits play a critical role in end-stage human heart failure. Here we report that the application of purified PP2AC significantly increased the Ca2+-sensitivity (ΔpCa50 = 0.05 ± 0.01) of the contractile apparatus in isolated skinned myocytes of non-failing (NF) hearts. A higher phosphorylation of troponin I (cTnI) was found at protein kinase A sites (Ser23/24) in NF compared to failing myocardium. The basal Ca2+-responsiveness of myofilaments was enhanced in myocytes of ischemic (ICM, ΔpCa50 = 0.10 ± 0.03) and dilated (DCM, ΔpCa50 = 0.06 ± 0.04) cardiomyopathy compared to NF. However, in contrast to NF myocytes the treatment with PP2AC did not shift force-pCa relationships in failing myocytes. The higher basal Ca2+-sensitivity in failing myocytes coincided with a reduced protein expression of PP2AC in left ventricular tissue from patients suffering from ICM and DCM (by 50 and 56% compared to NF, respectively). However, PP2A activity was unchanged in failing hearts despite an increase of both total PP and PP1 activity. The expression of PP2AB56α was also decreased by 51 and 62% in ICM and DCM compared to NF, respectively. The phosphorylation of cTnI at Ser23/24 was reduced by 66 and 49% in ICM and DCM compared to NF hearts, respectively. Our results demonstrate that PP2A increases myofilament Ca2+-sensitivity in NF human hearts, most likely via cTnI dephosphorylation. This effect is not present in failing hearts, probably due to the lower baseline cTnI phosphorylation in failing compared to non-failing hearts.
Similar content being viewed by others
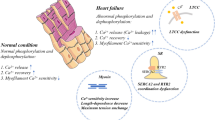
Introduction
The balance between protein kinases and opposing phosphatases is crucial for normal cell function in all tissues. Disruption of this balance between protein phosphorylation and dephosphorylation can trigger dysfunction and disease. Protein phosphatases are emerging as significant regulators in cellular signaling pathways. Protein phosphatase 2A (PP2A) has been identified as one important modulator of cellular processes, including regulation of ion channels and transporters, apoptosis, transcription, development, intercellular communication, and excitation–contraction coupling (Herzig and Neumann 2000; Janssens and Goris 2001).
This serine/threonine phosphatase consists of a heterotrimer that includes a dimeric core enzyme, containing a catalytic C subunit and a structural/scaffolding A subunit (Shi 2009). The core enzyme is bound to a variety of exchangeable regulatory B subunits, governing substrate specificity, subcellular targeting, and enzymatic activity (McCright et al. 1996; Price and Mumby 2000). One of the four unrelated gene families of the B subunit is B′ (or B56). Its members are encoded by five distinct genes, generating a variety of splicing isoforms, and are preferentially expressed in cardiac muscle (Ito et al. 2003; Zhou et al. 2007). Although there is growing knowledge about the structural properties of PP2A subunits, there is little information on their distribution and functional interplay in normal and diseased myocardium.
In the heart, PP2A is associated with the β-adrenergic receptor, connexin43, Na+/Ca2+ exchanger, L-type Ca2+ channel, ryanodine receptor (RyR), phospholamban (PLB), troponin I (cTnI), and myosin light chain 2 (MLC-2), representing critical molecules in cellular excitability and myocardial contractility (Ai and Pogwizd 2005; Chen et al. 2003; Deshmukh et al. 2007; Hall et al. 2006; Macdougall et al. 1991; Marx et al. 2000; Schulze et al. 2003). In intact myocytes localization of the PP2A regulatory B subunit at the sarcomeres is altered by activation of e.g. the β-adrenergic receptor (Deshmukh et al. 2007; Yin et al. 2010). The importance of PP2A targeting was tested in transgenic mice expressing a dominant negative mutant of the A subunit, which cannot bind regulatory B subunits, resulting in an altered sub-cellular localization of the enzyme, dilated cardiomyopathy, and depressed contractility (Brewis et al. 2000). The crucial role of PP2A in regulating cardiac Ca2+ signaling has been further demonstrated by application of the purified enzyme to isolated rat myocytes which was accompanied by a reduction of cytosolic Ca2+ transients (duBell et al. 1996). This is in line with data from a transgenic mouse model with heart-directed overexpression of PP2ACα showing a lower phosphorylation state of PLB and cTnI, hypertrophy, and impaired contractile function (Gergs et al. 2004). In the latter study (Gergs et al. 2004) increased diastolic Ca2+ levels were observed in cardiomyocytes from the PP2A-overexpressing transgenic mice, which may be due to the reduced phosphorylation of PLB and coincident reduced reuptake of Ca2+ into the sarcoplasmic reticulum (SR) via the SR Ca2+-ATPase. In contrast, application of PP2A inhibitors like cantharidin and calyculin A was associated with an increased phosphorylation of PLB, higher L-type Ca2+ currents and Ca2+ transients, and enhanced force of contraction (Boknik et al. 2001; duBell et al. 2002). These effects were independent of an increase in intracellular cAMP, an altered myofilament Ca2+-sensitivity, or K+ current inhibition (duBell et al. 2002; Neumann et al. 1994, 1995). Because a number of studies suggested a regulatory role of PP2A in normal myocardium and in animal models of the diseased heart, we hypothesized that the activity state of the PP2A multimeric complex is important for the progression of human heart failure.
Thus, the aim of the present study was to determine the functional relevance of PP2A by measuring the Ca2+-sensitivity of myofilaments in the presence and absence of PP2AC in isolated skinned myocytes of non-failing and failing human hearts. Moreover, we tested whether potential effects in myofilament Ca2+-sensitivity are paralleled by changes in PP2A expression and activity in failing hearts. Finally, we examined if differences in myofilament responsiveness to PP2A and in PP expression/activity correspond with the phosphorylation status of myofilament proteins in failing and non-failing human myocardium.
Methods
Human myocardial tissue
Right (RV) and left ventricular (LV) tissue was obtained from patients undergoing heart transplantation due to end-stage heart failure resulting from ischemic (ICM) or dilated cardiomyopathy (DCM) and from non-failing hearts (NF) that could not be transplanted due to medical reasons or blood group incompatibility (Table 1). Most patients received diuretics, cardiac glycosides, ACE inhibitors, and carvedilol (for detailed information see Table 1). Myocardial tissue was quickly transferred to cold cardioplegic solution and upon arrival in the laboratory, stored in liquid nitrogen. Care was taken not to use scarred, fibrotic, adipose samples, endocardium, epicardium, or great vessels. Tissue was frozen within 1 h of explantation. Permissions for the study were obtained from the local Ethic’s Committees of the University Hospital of Münster and the St Vincents’ Hospital Human Research Ethics Committee in Sydney, Australia (File number: H03/118; Title: Molecular Analysis of Human Heart Failure). The investigation conformed with the principles outlined in the Declaration of Helsinki.
Isolation and skinning of myocytes
Myocytes from LV of non-failing and failing hearts were mechanically isolated on ice as described previously (van der Velden et al. 1998). In the ICM group myocytes were isolated from the non-ischemic remodeled part of the left ventricle. Briefly, tissue was thawed in isolation solution containing 10 mmol/l imidazole (pH 7.0), 140 mmol/l KCl, 6 mmol/l Na2ATP, 2 mmol/l EGTA, and 6 mmol/l MgCl2. All membrane structures of isolated myocytes were dissolved by incubation with the isolation solution supplemented with 0.5% Triton X-100 for 5 min at 4°C. At this temperature the kinases and the phosphatases are inactive and therefore during myocyte isolation and skinning the phosphorylation status remains unchanged. In addition, after skinning all the soluble and membrane-bound protein kinases and phosphatases are washed away by washing three times with Triton-free isolation solution. The phosphorylation status of myofibrillar proteins is not affected by the procedure (Duncker et al. 2009; Kooij et al. 2010a, b; van der Velden et al. 2003a). Skinned myocytes were kept at 4°C up to 24 h.
Measurement of force-[Ca2+] relation in skinned myocytes
The use of skinned myocytes allows the study of myofibrillar contractility under standardized conditions, i.e. composition of intracellular buffer and sarcomere length. Measurement of isometric force was performed at 15°C and sarcomere length was set to 2.2 μm (van der Velden et al. 2003b). The composition of relaxing and activating solutions (pH 7.1) was calculated as described (Fabiato 1981). The pCa value, i.e. −log10[Ca2+], of the relaxing and activating solution were set to 9.0 and 4.5, respectively. Solutions with intermediate free [Ca2+] were obtained by appropriate mixing. Measurement of isometric force was started with a first activation of the myocyte at a pCa value of 4.5. The second activation was used to determine maximal isometric tension. The next measurements were performed at submaximal [Ca2+], followed by a final control measurement at maximal [Ca2+] (pCa = 4.5). Force values at submaximal [Ca2+] were normalized to interpolated control values, taking into account a linear reduction in maximal force with each activation. This force-[Ca2+] relationship was repeated after treatment with exogenous PP2A. For this purpose, myocytes were incubated for 40 min at 20°C in relaxing solution containing 5 U/ml of the active or heat-inactivated catalytic subunit (α/β) of PP2A (Promega). At this temperature PP2A is active and dephosphorylates its target proteins. PP2A was not present in the solutions used during the tension-pCa measurements. Incubation for 40 min at 20°C in relaxing solution without PP2A did not change phosphorylation status of the myofilament proteins (unpublished data). Force-[Ca2+] relations were fit to a modified Hill equation (van der Velden et al. 2003a). The maximal rate of force redevelopment (max ktr) was determined from an exponential curve fit of force redevelopment after a slack test in activating solution with pCa 4.5.
Immunological detection of PP2A subunits
Frozen RV and LV tissue of human hearts was homogenized at 4°C for 90 s in a buffer containing 20 mmol/l Tris/HCl (pH 7.4), 1 mmol/l EDTA, 5 mmol/l MgCl2, 1 mmol/l DTT, and protease inhibitors. After incubation for 20 min on ice samples were centrifuged at 15,800 × g for 15 min. Supernatants were diluted in 5% SDS buffer containing 62.5 mmol/l Tris/HCl (pH 6.8), 5% glycerol, and 40 mmol/l DTT. For immunoblot analysis different amounts of solubilized proteins were separated electrophoretically in 10% SDS–polyacrylamide gels. Proteins were transferred to nitrocellulose for immunoblotting. Nitrocellulose sheets were probed with different antibodies: polyclonal anti-PP2ACα/β antibody (Santa Cruz Biotechnology), a polyclonal anti-Aα antibody (Santa Cruz), a polyclonal anti-B56α antibody (Santa Cruz), and a polyclonal anti-calsequestrin antibody (Kirchhefer et al. 2001). Antibody binding was visualized by alkaline phosphatase-conjugated secondary antibodies (Amersham Biosciences) or by 125I-labeled protein A (PerkinElmer) followed by autoradiography. Labeling intensities were quantified with use of a PhosphorImager.
Analysis of the phosphorylation status of myofilament proteins
LV samples were TCA(trichloroacetic acid)-treated as described previously (Zaremba et al. 2007). Samples were separated on a gradient gel (Criterion Tris–HCl 4–15% gel, BioRad) and proteins were stained for 1 h with ProQ Diamond Phosphoprotein Stain (Molecular Probes) (Hamdani et al. 2010). Staining was visualized using the LAS-3000 Image Reader (FUJI; 460 nm/605 nm Ex/Em; 2 min illumination) and signals were analyzed with AIDA. Subsequently, gels were stained overnight with SYPRO Ruby stain (Molecular Probes) and visualized with the LAS-3000. ProQ-stained phosphorylated proteins were normalized to SYPRO Ruby-stained myosin binding protein C (cMyBP-C) to correct for differences in sample loading. Gel electrophoresis and Western blotting was performed to analyze bisphosphorylation of cTnI at PKA sites Ser23/24 (rabbit polyclonal antibody, Cell Signaling; dilution 1:500). Signals were normalized to actin to correct for differences in protein loading.
Incubation of myofilaments with PP2A
To assess the effect of PP2A on myofilament protein phosphorylation, NF myocardium was isolated and skinned as described above (as for force measurements) and incubated in relaxing solution with 0.5 U or 5 U exogenous PP2AC for 40 min at RT. In the control incubation no PP2A was added to NF tissue. After incubation, protein was isolated via the 2-D Clean-Up Kit (GE Healthcare) and dissolved in 1D-sample buffer containing 15% glycerol (v/v), 62.5 mM Tris (pH 6.8), 1% (w/v) SDS and 2% (w/v) DTT. To determine the phosphorylation status of the myofilament proteins ProQ Diamond Phosphoprotein Stain was used. The amount of phosphorylation of cTnI-Ser23/24 was studied via Western blotting.
Protein phosphatase assay
PP activity was determined with [32P]-phosphorylase a as substrate (Boknik et al. 2000). Portions of pulverized frozen LV tissue were homogenized at 4°C for 3 × 30 s each with a Polytron in a buffer containing 4 mmol/l EDTA and 15 mmol/l β-mercaptoethanol (pH 7.4). The homogenate was centrifuged for 20 min at 14,000 × g. The incubation mixture contained 20 mmol/l Tris/HCl (pH 7.0), 5 mmol/l caffeine, 0.1 mmol/l EDTA, and 0.1% β-mercaptoethanol (vol/vol). The reaction was started by adding aliquots of supernatants, and 3 nmol/l okadaic acid was used to differentiate between PP1 and PP2A activity. The reaction was stopped by addition of 50% TCA. Precipitated protein was sedimented by centrifugation, and the radioactivity was counted in the supernatant.
Statistics
Data are reported as means ± SEM. Statistical differences between groups were calculated by ANOVA followed by Bonferroni’s t-test. P < 0.05 was considered significant.
Results
PP2A increases force development at submaximal [Ca2+] in NF myofilaments
To investigate whether the dephosphorylation of contractile proteins by PP2A directly influences isometric force development, we applied the catalytic subunit of the enzyme to skinned myocytes from NF hearts. For this purpose, single myocytes were attached between a force transducer and a piezoelectric motor (Fig. 1a). All experiments were performed on the stage of an inverted microscope. In Fig. 1b representative recordings of force development before and after application of PP2AC at saturating (pCa = 4.5) [Ca2+] are shown. Treatment of myocytes with PP2AC did not change maximal or passive isometric force (Table 2). Representative recordings of force development at submaximal (pCa values ranged from 5.0 to 6.0; here pCa = 5.4) [Ca2+] are shown in Fig. 1b and demonstrate a higher force development after PP2A treatment indicating that Ca2+-sensitivity is modulated by the enzyme in NF myocytes. Indeed, the measurement of the force-pCa relationship (Fig. 1c) revealed a higher Ca2+-responsiveness of the myofilaments after incubation with PP2AC as evidenced by an enhanced pCa50 value (ΔpCa50 = 0.05 ± 0.01). Incubation of skinned myocytes with heat-inactivated PP2A did not shift force-pCa relationship demonstrating an enzyme-specific effect (data not shown).

The image shows a single NF myocyte in relaxing solution attached between a force transducer and a piezoelectric motor (a). Shown are original recordings (b) of isometric force development in a Triton-skinned NF myocyte before and after treatment with PP2AC. The force was recorded at a sarcomere length of 2.2 μm during maximal activation at pCa = 4.5 and submaximal activation at pCa = 5.4. Force increases when the myocyte is transferred from the relaxing solution (pCa = 9.0) into the activating solution. After reaching steady state force, the cardiomyocyte was 20% reduced in length within 2 ms and re-stretched after 30 ms (slack test). During this slack test, force first dropped to zero and after the re-stretch quickly redeveloped to the original steady state level. Subsequently, the cell was transferred back to relaxing solution and a second slack test was started to determine passive force. The active force of a myocyte is the steady state force (maximal force) minus the passive force. The summarized force-pCa relationship is given (c; 6 NF, 14 cells)
Higher basal Ca2+ sensitivity in ischemic heart failure but no effect by PP2A
Ca2+-sensitivity was also determined before (Fig. 2a) and after (Fig. 2b) PP2AC application in failing myocytes by measuring force in activating solutions with submaximal [Ca2+] obtained by appropriate mixing of the activating and relaxation solutions. After maximal activation (pCa = 4.5) six measurements were carried out at submaximal [Ca2+] followed by a maximal activation. Cells were excluded from the data when the active force dropped more than 20% between the first and the last measurement at pCa = 4.5 before PP2AC treatment of the cells. In ICM myocyte preparations, the basal Ca2+-sensitivity was significantly higher compared to NF (ΔpCa50 = 0.10 ± 0.03) (Fig. 2c). However, PP2AC treatment did not further shift force-pCa relationship (ΔpCa50 = 0.01 ± 0.01) suggesting maximal dephosphorylation in ICM myocytes. In DCM myocardium, basal Ca2+-sensitivity was also higher compared to NF (ΔpCa50 = 0.06 ± 0.04), however not significantly (Fig. 2c). The application of PP2AC did not change the Ca2+-responsiveness of myofilaments compared to untreated DCM myocytes (ΔpCa50 = 0.01 ± 0.01).

Isometric force was determined at different [Ca2+] before (a) and after (b) treatment with PP2AC in NF (6 NF, 14 cells) and failing (ICM, 6 patients, 14 cells; DCM, 6 patients, 13 cells) myocytes. Force at submaximal [Ca2+] was normalized to the force at saturating [Ca2+] (pCa = 4.5). PP2AC treatment increased Ca2+-sensitivity only in NF cardiomyocytes (c). Basal Ca2+-sensitivity was higher in ICM compared to NF. **P < 0.01 before vs. after PP2A treatment (Repeated measures Two-Way ANOVA, n = 6); #P < 0.05 vs. NF (Two-Way ANOVA (Source of Variation: patient) NS, One-way ANOVA (Ca2+-sensitivity NF, ICM, DCM before PP2A) P = 0.06, Bonferroni’s Multiple comparison test NF vs ICM P < 0.05, n = 6)
PP2A does not effect the steepness of the force-pCa curves and rate of tension redevelopment
The steepness of the force-pCa curves (nH) and the maximal rate of tension redevelopment (max ktr) were also determined before and after PP2AC treatment. Both nH and max ktr were not significantly affected by PP2A in NF, ICM and DCM cardiomyocytes (Table 2).
PP2A dephosphorylates cTnI in donor cardiomyocytes
NF heart tissue was incubated with 0.5 U or 5 U PP2AC to test which myofilament proteins were dephosphorylated by PP2A. Via SYPRO Ruby and ProQ Diamond stainings of gels it was demonstrated that PP2A dephosphorylates cTnI (Fig. 3a). The phosphorylation level of other myofilament proteins (cMyBP-C, troponin T, Desmin and MLC-2) was not affected (data not shown). Western blotting with an antibody raised against bis-phosphorylated cTnI at PKA sites Ser23/24 confirmed that cTnI was dephosphorylated by PP2A at the PKA sites (Fig. 3b).

NF myocardium (n = 2) was treated with 0U, 0.5U or 5U PP2A for 40 min at RT. Subsequently, proteins were separated by 1D gel electrophoresis (a). Phosphorylation signals of all proteins on ProQ Diamond-stained gels were divided by their SYPRO signals to correct for minor differences in loading. PKA-dependent bis-phosphorylation of cTnI at Ser23/24 was measured by Western blot analysis and normalized to α-actinin (b). cTnT troponin T, MLC-2 myosin light chain 2, MHC myosin heavy chain, MLC-1 myosin light chain 1, MW molecular weight marker
Reduced PKA-dependent cTnI phosphorylation in human heart failure
To study whether an altered basal phosphorylation level of myofilament proteins is responsible for the higher basal Ca2+-responsiveness in failing hearts, we performed SYPRO Ruby and ProQ Diamond stainings of gels loaded with NF, ICM and DCM LV samples (Fig. 4a). Total phosphorylation of cTnI was lower in failing samples compared to NF hearts (Fig. 4b). No differences were observed in the phosphorylation status of the other myofilament proteins on the gel. The phosphorylation level of cTnI at PKA-sites was studied by Western blotting using a phosphospecific antibody against bis-phosphorylated Ser23/24 of cTnI (Fig. 4c). Here we measured a lower PKA-dependent phosphorylation of cTnI in ICM and DCM hearts compared to NF tissue.

Myofilament proteins from NF and failing (ICM; DCM) hearts were separated by 1D gel electrophoresis (a). Staining differences between gels were corrected with phosphorylated ovalbumin of the peppermint marker (PM). Phosphorylation signals of all proteins on ProQ Diamond-stained gels were divided by the SYPRO signals of myosin binding protein C (cMyBP-C) to correct for minor differences in loading (b). PKA-dependent bis-phosphorylation of cTnI at Ser23/24 was measured by Western blot analysis and normalized to actin (c). cTnT troponin T, MLC-2 myosin light chain 2, MW molecular weight marker. *P < 0.05 vs. NF (post-hoc Bonferroni analysis)
Reduced expression of PP2A subunits Cα/β and B56α in failing hearts
To test whether PP2A may be involved in the higher basal myofilament Ca2+-sensitivity of failing human hearts, we determined the expression level of major PP2A subunits in heart samples from patients suffering from ischemic and dilated cardiomyopathy. The catalytic subunit C of PP2A was detected as a 36-kDa protein in cardiac homogenates (Fig. 5a). In RV tissue, PP2AC protein expression was unchanged between all groups investigated, whereas it was reduced by 50 and 56% in LV of ICM and DCM, respectively, compared to NF hearts. Moreover, we found that the expression of B56α was decreased in RV tissue by 87% in DCM but not in ICM hearts compared to NF (Fig. 5b). In contrast, the expression of B56α was reduced in LV homogenates of both ICM (by 51%) and DCM (by 62%) hearts. All samples were also probed with an antibody recognizing the structural subunit of PP2A, Aα. This subunit runs at 65 kDa on SDS–PAGE (Fig. 5c). Here, we measured a similar protein expression of Aα in ventricles of ICM, DCM, and NF hearts. Linearity of signals detected by immunoblot analysis of heart samples was confirmed (data not shown). Signals were standardized to the expression of calsequestrin, acting as the main SR Ca2+ storage protein. No difference in the protein expression of calsequestrin was detected between failing and non-failing hearts (data not shown). This is in agreement with data from other heart failure models (Phillips et al. 1998).

Homogenates from NF and failing (ICM; DCM) RV and LV myocardium were solubilized in a SDS-buffer and then separated electrophoretically. The immunological detection of PP2A subunits was performed by use of specific antibodies directed against Cα/β (a), B56α (b), and Aα (c). The statistical summarization of quantification is depicted as PhosphorImager units. Signals were standardized to calsequestrin (CSQ). *P < 0.05 vs. NF; + P < 0.05 vs. RV
Unchanged PP2A but increased PP1 activity in ICM hearts
The measurement of PP activity was used to determine whether the lower PP2AC expression in failing LV myocardium is paralleled by comparable changes of the catalytic activity. Total phosphatase activity was enhanced by 35% in ICM compared to non-failing homogenates, whereas no difference was observed between DCM and NF (Fig. 6a). To discriminate PP2A activity from PP1 activity, we added 3 nmol/l okadaic acid (OA) to the reaction mixture, a concentration known to inhibit only PP2A but not PP1 (Cohen 1991). Under our experimental conditions (phosphorylase a as substrate, absence of divalent cations) only PP1 and PP2A is measurable. Therefore, PP2A activity was determined as total activity minus activity in the presence of 3 nmol/l OA. The contribution of PP2A to the total PP activity was shifted from 52% in non-failing hearts to 41% in ICM hearts. However, total PP2A activity was unchanged between ICM and NF preparations (Fig. 6b). The higher total phosphatase activity in ICM reflects an increase in PP1 activity. In DCM hearts, PP2A and PP1 activities were unchanged compared to NF samples (Fig. 6b).

Total PP activity was determined in supernatants from NF and failing (ICM; DCM) hearts using [32P]-labeled phosphorylase a as substrate (a). Aliquots of supernatants were also assayed in the presence of 3 nmol/l okadaic acid, representing the concentration that inhibits only PP2A but not PP1. Therefore, PP2A activity was calculated as the total PP activity minus the activity that was not inhibited by okadaic acid (b). *P < 0.05 vs. NF
Discussion
In the present study, we tested whether PP2A is involved in the regulation of myofilament contractility in human Triton-permeabilized cardiomyocytes. The application of PP2AC resulted in a higher Ca2+-sensitivity and dephosphorylation (at PKA sites) of cTnI in non-failing LV myocardium indicating that PP2A may modulate myofilament function by dephosphorylation of cTnI. PP2AC treatment of non-failing myocytes changed the cTnI phosphorylation level as well as the myofilament Ca2+-sensitivity to a level intermediate between the baseline levels observed in non-failing myocytes and failing myocytes. This finding is in line with studies showing cTnI as a main target of PP2A (Deshmukh et al. 2007; Yin et al. 2010). Our data are in contrast to control myocytes of whole rat hearts where the addition of PP2AC had no effect on myofilament Ca2+-sensitivity (Belin et al. 2007). This suggests that PP2A is more effective in human than in rodent myocardium. It is conceivable that differences in the developmental regulation are responsible for the functional differences between both species. For example, mRNA expression, protein levels, and activity of PP2A were lower in adult compared to embryonic rat hearts suggesting a limited role of the enzyme in regulating myofilament contractility in rat heart (Gombosova et al. 1998; Heller et al. 1998).
Next we measured the basal Ca2+-sensitivity of force in failing myocytes. The higher Ca2+-responsiveness in failing compared to NF myocytes is in accordance with previous studies on failing human hearts (van der Velden et al. 2003a; van der Velden et al. 2003b; Wolff et al. 1996). A higher Ca2+-sensitivity in failing hearts has been ascribed to a reduction in the PKA-mediated phosphorylation of cardiac myofilament proteins (Wolff et al. 1996). Indeed, we detected a diminished PKA-dependent cTnI phosphorylation in failing myocardium. This may be caused by a depressed β-adrenergic signaling characterized by a decreased β-adrenergic receptor density, a higher Gi expression, and reduced cAMP levels (Feldman 1993). Consistent with this, the application of PKA abolished the increased Ca2+-sensitivity of force in failing human myocytes leading to values observed in non-failing cells in the absence of the enzyme (van der Velden et al. 2003b). In the latter study a positive correlation between Ca2+-sensitivity and dephosphorylated cTnI was observed. On the other hand, Ca2+-sensitivity is not only dependent on kinase (PKA)-mediated phosphorylation of myofilament proteins but also on the dephosphorylation by PP’s. In the present study, we found an unchanged PP2A and a higher PP1 activity in failing hearts. Incubation of non-failing human myocytes with PP1 has been associated with a reduced Ca2+-sensitivity of force and hastened relaxation. These effects were explained by a dephosphorylation of MLC-2 (van der Velden et al. 2003a). It is conceivable that the desensitization by a PP1-dependent MLC-2 dephosphorylation was overcome by the effect of cTnI dephosphorylation resulting in an overall higher basal Ca2+-responsiveness of force in failing samples. In contrast to non-failing myocytes, PP2AC administration had no effect on Ca2+-sensitivity in failing myocytes. In agreement with our data, the addition of purified PP2AC had no effects on Ca2+ sensitivity in a rat model of congestive heart failure (Belin et al. 2007). PP2A-dependent dephosphorylation of native thin filaments isolated from human ICM and DCM ventricles was also associated with an unchanged Ca2+-sensitivity (Noguchi et al. 2004). These data suggest that PP2A does not exert a functional effect in failing hearts due to the low cTnI phosphorylation level.
The functional effects of exogenously applied PP2AC on Ca2+-sensitivity of force may also depend on the expression of the enzyme and its subunits. Here, the reduced PP2AC expression in failing LV may have significant consequences for myofilament contraction since Cα was identified as the predominant isoform in human myocardium (Luss et al. 2000). In animal models, an increased PP2A activity was measured in rat hearts after long-term β-adrenergic stimulation, resulting in a compensated hypertrophy (Boknik et al. 2000). A higher PP2A expression and activity was also detected in hearts of mice with primary pulmonary hypertension (Larsen et al. 2008). In a dog model of acute myocardial infarction, PP2A expression was also enhanced 5 days post occlusion in the border zone (Hund et al. 2009). In contrast, Gupta and co-workers (2003) failed to detect any differences of PP2A activity in microembolized dog hearts, resulting in chronic heart failure. Therefore, it was surprising that the PP2AC expression was reduced in failing human hearts. It appears that an acute myocardial stress by either mechanical overload (e.g. induced by β-adrenergic stimulation) or phases of ischemia is associated with an increase of the catalytic subunit, whereas in decompensated heart failure PP2AC expression is decreased. It is also conceivable that the lower PP2AC expression may compensate for the reduced PKA-dependent phosphorylation of cTnI or the enhanced PP1 activity in ICM heart homogenates. A higher PP1 activity was also observed in SR membrane preparations of failing human DCM hearts, whereas no significant difference between PP activities in homogenates was noted (Neumann et al. 1997), as observed in the present study. The detrimental effects of an increased PP1 activity to cardiac contractility were demonstrated in transgenic mice (Carr et al. 2002). Beyond this simplified model of keeping a cellular homeostasis between the main cardiac PP isoforms, the lower expression of PP2AC in ICM might help to protect the heart tissue against ischemic damages. In this context, fostriecin, an inhibitor of PP2A, limited myocardial infarct size in isolated rabbit hearts when administered after the onset of ischemia (Weinbrenner et al. 1998), whereas transgenic mice with cardiac-specific overexpression of PP2ACα exhibited isles of necrosis, fibrosis, and contractile failure (Gergs et al. 2004).
Interestingly, PP2AC expression was decreased in LV but not in RV chambers of failing hearts. Data on protein expression comparing different chambers of failing hearts are rare. It has been shown that the expression levels of PP2A-associated connexin43 (Ai and Pogwizd 2005; Dupont et al. 2001) and L-type Ca2+ channel (Gruver et al. 1994) are reduced in LV of failing human hearts, indicating a predominant affection of the LV function in heart failure. It remains to be tested whether the lower expression of PP2AC compensates for the reduction of both regulatory proteins in failing LV. Alternatively, the inhibition of PP2AC has been shown to minimize the metabolic damage of LV tissue in failing myocardium (Weinbrenner et al. 1998).
In the present study, the lower PP2AC expression was not followed by a corresponding change in activity. This discrepancy may depend on the subcellular distribution of the enzyme which can be modulated by the expression of its regulatory subunits (e.g. B56α). In failing human hearts, we have demonstrated a concomitantly reduced expression of PP2AB56α. As reported above, B56α is thought to be responsible for the subcellular targeting of the PP2A dimeric core enzyme to specific substrates (Depaoliroach et al. 1994). A marked loss in B56α protein expression was also detected in rat neonatal myocytes after activation of JNK, a stress-related stimulus in the manifestation of cardiac hypertrophy and failure (Glaser et al. 2006). A decrease in PP2AB56α expression was also measured in a model of sepsis-associated cardiac dysfunction (Marshall et al. 2009). The down-regulation was associated with a lower expression of PP2AC and increased cTnI phosphorylation levels. This supports the hypothesis that a reduced PP2AB56α expression may impair the targeting of PP2AC to its subcellular sites. Indeed, decreased levels of RyR-bound PP2AC were detected in failing human hearts (Marx et al. 2000). This may contribute to a hyperphosphorylation of the channel, resulting in a defective regulation of excitation–contraction coupling. Here, we detected higher expression levels of PP2AB56α in LV compared to RV of non-failing and failing hearts. These differences may point to localized responses to cardiac overload. For instance, βMHC transcript levels were lower in LV suggesting that there is more need for an increase in contractility in LV compared to RV (Sharma et al. 2003). Consistently, LV exhibits a higher RyR expression (Munch et al. 2000) which enables a stronger SR Ca2+ release and high contractile velocity of myofibrils. We suggest that a higher PP2AB56α expression in LV may help to target the catalytic subunit to the more abundant RyR.
Our data indicate that PP2A may be involved in the regulation of the myofilament Ca2+-sensitivity in non-failing human hearts. In contrast, PP2A had no effect on the enhanced basal Ca2+-sensitivity in failing myocytes which might result from the depressed PKA-dependent basal phosphorylation of cTnI in failing hearts. The lower expression of the catalytic subunit and B56α in ICM might be compensatory mechanisms in order to normalize the higher basal Ca2+-sensitivity.
References
Ai X, Pogwizd SM (2005) Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ Res 96:54–63
Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, de Tombe PP (2007) Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res 101:195–204
Boknik P, Fockenbrock M, Herzig S, Knapp J, Linck B, Luss H, Muller FU, Muller T, Schmitz W, Schroder F, Neumann J (2000) Protein phosphatase activity is increased in a rat model of long-term beta-adrenergic stimulation. Naunyn Schmiedebergs Arch Pharmacol 362:222–231
Boknik P, Khorchidi S, Bodor GS, Huke S, Knapp J, Linck B, Luss H, Muller FU, Schmitz W, Neumann J (2001) Role of protein phosphatases in regulation of cardiac inotropy and relaxation. Am J Physiol Heart Circ Physiol 280:H786–H794
Brewis N, Ohst K, Fields K, Rapacciuolo A, Chou D, Bloor C, Dillmann W, Rockman H, Walter G (2000) Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am J Physiol Heart Circ Physiol 279:H1307–H1318
Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, Depaoli-Roach AA, Kranias EG (2002) Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol 22:4124–4135
Chen Y, Rajashree R, Liu QH, Hofmann P (2003) Acute p38 MAPK activation decreases force development in ventricular myocytes. Am J Physiol Heart Circ Physiol 285:H2578–H2586
Cohen P (1991) Classification of protein-serine threonine phosphatases—identification and quantitation in cell extracts. Methods Enzymol 201:389–398
Depaoliroach AA, Park IK, Cerovsky V, Csortos C, Durbin SD, Kuntz MJ, Sitikov A, Tang PM, Verin A, Zolnierowicz S (1994) Serine/threonine protein phosphatases in the control of cell function. Adv Enzyme Regul 34:199–224
Deshmukh PA, Blunt BC, Hofmann PA (2007) Acute modulation of PP2a and troponin I phosphorylation in ventricular myocytes: studies with a novel PP2a peptide inhibitor. Am J Physiol Heart Circ Physiol 292:H792–H799
DuBell WH, Lederer WJ, Rogers TB (1996) Dynamic modulation of excitation–contraction coupling by protein phosphatases in rat ventricular myocytes. J Physiol 493:793–800
DuBell WH, Gigena MS, Guatimosim S, Long XL, Lederer WJ, Rogers TB (2002) Effects of PP1/PP2A inhibitor calyculin A on the E-C coupling cascade in murine ventricular myocytes. Am J Physiol Heart Circ Physiol 282:H38–H48
Duncker DJ, Boontje NM, Merkus D, Versteilen A, Krysiak J, Mearini G, El-Armouche A, de Beer VJ, Lamers JMJ, Carrier L, Walker LA, Linke WA, Stienen GJM, van der Velden J (2009) Prevention of myofilament dysfunction by beta-blocker therapy in post-infarct remodeling. Circ Heart Failure 2:233–242
Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N, Kaprielian R, Yacoub MH, Severs NJ (2001) Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol 33:359–371
Fabiato A (1981) Myoplasmic free calcium-concentration reached during the twitch of an intact isolated cardiac cell and during calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned cardiac cell from the adult rat or rabbit ventricle. J Gen Physiol 78:457–497
Feldman AM (1993) Modulation of adrenergic receptors and G-transduction proteins in failing human ventricular myocardium. Circulation 87:27–34
Gergs U, Boknik P, Buchwalow I, Fabritz L, Matus M, Justus I, Hanske G, Schmitz W, Neumann J (2004) Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J Biol Chem 279:40827–40834
Glaser ND, Lukyanenko YO, Wang YB, Wilson GM, Rogers TB (2006) JNK activation decreases PP2A regulatory subunit B56 alpha expression and mRNA stability and increases AUF1 expression in cardiomyocytes. Am J Physiol Heart Circ Physiol 291:H1183–H1192
Gombosova I, Boknik P, Kirchhefer U, Knapp J, Luss H, Muller FU, Muller T, Vahlensieck U, Schmitz W, Bodor GS, Neumann J (1998) Postnatal changes in contractile time parameters, calcium regulatory proteins, and phosphatases. Am J Physiol Heart Circ Physiol 274:H2123–H2132
Gruver EJ, Morgan JP, Stambler BS, Gwathmey JK (1994) Uniformity of calcium channel number and isometric contraction in human right and left ventricular myocardium. Basic Res Cardiol 89:139–148
Gupta RC, Mishra S, Rastogi S, Imai M, Habib O, Sabbah HN (2003) Cardiac SR-coupled PP1 activity and expression are increased and inhibitor 1 protein expression is decreased in failing hearts. Am J Physiol Heart Circ Physiol 285:H2373–H2381
Hall DD, Feekes JA, Don ASA, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC, Hell JW (2006) Binding of protein phosphatase 2A to the L-type calcium channel Ca(v)1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 45:3448–3459
Hamdani N, Borbely A, Veenstra SPGR, Zaremba R, dos Remedios C, Niessen HWM, Paulus WJ, Stienen GJM, van der Velden J (2010) Diverse alterations in sarcomeric protein composition and function in ischemic and idiopathic dilated cardiomyopathy. J Muscle Res Cell Motil 31:289–301
Heller FA, Xue C, Fisher A, Everett AD (1998) Expression and mapping of protein phosphatase 2A(alpha) in the developing rat heart. Pediatr Res 43:68–76
Herzig S, Neumann J (2000) Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev 80:173–210
Hund TJ, Wright PJ, Dun W, Snyder JS, Boyden PA, Mohler PJ (2009) Regulation of the ankyrin-B-based targeting pathway following myocardial infarction. Cardiovasc Res 81:742–749
Ito A, Koma YI, Sohda M, Watabe K, Nagano T, Misumi Y, Nojima H, Kitamura Y (2003) Localization of the PP2A B56 gamma regulatory subunit at the Golgi complex—possible role in vesicle transport and migration. Am J Pathol 162:479–489
Janssens V, Goris J (2001) Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353:417–439
Kirchhefer U, Neumann J, Baba HA, Begrow F, Kobayashi YM, Reinke U, Schmitz W, Jones LR (2001) Cardiac hypertrophy and impaired relaxation in transgenic mice overexpressing triadin 1. J Biol Chem 276:4142–4149
Kooij V, Boontje NM, Zaremba R, Jaquet K, dos Remedios C, Stienen GJM, van der Velden J (2010a) Protein kinase C α and ε phosphorylation of troponin and myosin binding protein C reduce Ca2+-sensitivity in human myocardium. Basic Res Cardiol 105:289–300
Kooij V, Saes M, Jaquet K, Zaremba R, Foster DB, Murphy AM, dos Remedios C, van der Velden J, Stienen GJM (2010b) Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background. J Mol Cell Cardiol 48:954–963
Larsen KO, Lygren B, Sjaastad I, Krobert KA, Arnkvaern K, Florholmen G, Larsen AKR, Levy FO, Tasken K, Skjonsberg OH, Christensen G (2008) Diastolic dysfunction in alveolar hypoxia: a role for interleukin-18-mediated increase in protein phosphatase 2A. Cardiovasc Res 80:47–54
Luss H, Klein-Wiele O, Boknik P, Herzig S, Knapp J, Linck B, Muller FU, Scheld HH, Schmid C, Schmitz W, Neumann J (2000) Regional expression of protein phosphatase type 1 and 2A catalytic subunit isoforms in the human heart. J Mol Cell Cardiol 32:2349–2359
Macdougall LK, Jones LR, Cohen P (1991) (Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur J Biochem 196:725–734
Marshall M, Anilkumar N, Layland J, Walker SJ, Kentish JC, Shah AM, Cave AC (2009) Protein phosphatase 2A contributes to the cardiac dysfunction induced by endotoxemia. Cardiovasc Res 82:67–76
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101:365–376
McCright B, Rivers AM, Audlin S, Virshup DM (1996) The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem 271:22081–22089
Munch G, Bolck B, Sugaru A, Schwinger RHG (2000) Isoform expression of the sarcoplasmic reticulum Ca2+ release channel (ryanodine channel) in human myocardium. J Mol Med 78:352–360
Neumann J, Boknik P, Herzig S, Schmitz W, Scholz H, Wiechen K, Zimmermann N (1994) Biochemical and electrophysiological mechanisms of the positive inotropic effect of calyculin-A, a protein phosphatase inhibitor. J Pharmacol Exp Ther 271:535–541
Neumann J, Herzig S, Boknik P, Apel M, Kaspareit G, Schmitz W, Scholz H, Tepel M, Zimmermann N (1995) On the cardiac contractile, biochemical and electrophysiological effects of cantharidin, a phosphatase inhibitor. J Pharmacol Exp Ther 274:530–539
Neumann J, Eschenhagen T, Jones LR, Linck B, Schmitz W, Scholz H, Zimmermann N (1997) Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol 29:265–272
Noguchi T, Hunlich M, Camp PC, Begin KJ, El Zaru M, Patten R, Leavitt BJ, Ittleman FP, Alpert NR, LeWinter MM, VanBuren P (2004) Thin filament-based modulation of contractile performance in human heart failure. Circulation 110:982–987
Phillips RM, Narayan P, Gomez AM, Dilly K, Jones LR, Lederer WJ, Altschuld RA (1998) Sarcoplasmic reticulum in heart failure: central player or bystander? Cardiovasc Res 37:346–351
Price NE, Mumby MC (2000) Effects of regulatory subunits on the kinetics of protein phosphatase 2A. Biochemistry 39:11312–11318
Schulze DH, Muqhal M, Lederer WJ, Ruknudin AM (2003) Sodium/calcium exchanger (NCX1) macromolecular complex. J Biol Chem 278:28849–28855
Sharma S, Razeghi P, Shakir A, Keneson BJ, Clubbb F, Taegtmeyer H (2003) Regional heterogeneity in gene expression profiles: a transcript analysis in human and rat heart. Cardiology 100:73–79
Shi YG (2009) Serine/threonine phosphatases: mechanism through structure. Cell 139:468–484
van der Velden J, Klein LJ, van der Bijl M, Huybregts MAJM, Stooker W, Witkop J, Eijsman L, Visser CA, Visser FC, Stienen GJM (1998) Force production in mechanically isolated cardiac myocytes from human ventricular muscle tissue. Cardiovasc Res 38:414–423
van der Velden J, Papp Z, Boontje NM, Zaremba R, de Jong JW, Janssen PML, Hasenfuss G, Stienen GJM (2003a) The effect of myosin light chain 2 dephosphorylation on Ca2+-sensitivity of force is enhanced in failing human hearts. Cardiovasc Res 57:505–514
van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PBJ, Goldmann P, Jaquet K, Stienen GJM (2003b) Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res 57:37–47
Weinbrenner C, Baines CP, Liu GS, Armstrong SC, Ganote CE, Walsh AH, Honkanen RE, Cohen MV, Downey JM (1998) Fostriecin, an inhibitor of protein phosphatase 2A, limits myocardial infarct size even when administered after onset of ischemia. Circulation 98:899–905
Wolff MR, Buck SH, Stoker SW, Greaser ML, Mentzer RM (1996) Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies—role of altered beta-adrenergically mediated protein phosphorylation. J Clin Invest 98:167–176
Yin X, Cuello F, Mayr U, Hao Z, Hornshaw M, Ehler E, Avkiran M, Mayr M (2010) Proteomics analysis of the cardiac myofilament subproteome reveals dynamic alterations in phosphatase subunit distribution. Mol Cell Proteomics 9:497–509
Zaremba R, Merkus D, Hamdani N, Lamers JMJ, Paulus WJ, dos Remedios C, Duncker DJ, Stienen GJM, van der Velden J (2007) Quantitative analysis of myofilament protein phosphorylation in small cardiac biopsies. Proteomics Clin Appl 1:1285–1290
Zhou XW, Mudannayake M, Green M, Gigena MS, Wang GH, Shen RF, Rogers TB (2007) Proteomic studies of PP2A-B56 gamma 1 phosphatase complexes reveal phosphorylation-regulated partners in cardiac local signaling. J Proteome Res 6:3433–3442
Acknowledgments
The study was supported by the IMF Münster (to U.K.) and the Deutsche Forschungsgemeinschaft (BO1263/9-1 and NE393/32-1).
Disclosures
No conflicts of interest are declared by the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wijnker, P.J.M., Boknik, P., Gergs, U. et al. Protein phosphatase 2A affects myofilament contractility in non-failing but not in failing human myocardium. J Muscle Res Cell Motil 32, 221–233 (2011). https://doi.org/10.1007/s10974-011-9261-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-011-9261-x