
Abstract
The paper describes the age (production date) determination of uranium reference materials using the 231Pa/235U ratio. Direct addition of 237Np in secular equilibrium with its 233Pa daughter was chosen instead of the regular milking of 237Np to avoid possible loss of Pa. Sample preparation consists of a fast, one-step procedure. The developed method using ICP-MS for the measurement of 231Pa is more precise than alpha spectrometry and is applicable for freshly produced low-enriched uranium materials. The measured ages are in good agreement with the reported production dates, thus the 231Pa/235U chronometer can be applied for validation of 230Th/234U in nuclear forensics and safeguards.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nuclear materials are strictly controlled by the nuclear safeguards regimes. If such material, however, gets out of the regulatory control and is confiscated afterwards, a detailed examination should be performed to identify the intended use, origin and last legal owner of the material [1, 2]. Nuclear forensic analysis uses several signatures, like U or Pu isotopic composition, fuel pellet dimensions, chemical form and impurities, isotope ratios of minor constituents such as S, Sr, Nd, and Pb, to provide hints on the production history of the material and to narrow down the possible facilities being in connection with the material [1,2,3,4]. One of the nuclear forensic signatures is the time elapsed since the last chemical or physical purification of the material, commonly called the age of the material, can be measured for radioactive, and thus also for nuclear materials [1, 5,6,7]. This unique opportunity is based on exploiting the presence and decay of radionuclides: when radioactive material is chemically or physically purified from the impurities, also the radioactive decay products are separated. After this separation, the radioactive progenies start to grow-in into the material. By measuring the daughter-to-parent ratio in the sample, the time elapsed since the last separation can be calculated according to the decay equations (Bateman-equations), when assuming that the parent-daughter separation was complete during the process. In contrast to most other nuclear forensic signatures, the production date of the material is a predictive signature, thus it does not require databases or comparison samples for interpretation (i.e. it is a self-explaining signature). This feature makes the age of the material one of the most prominent signatures for attribution.
The age can be calculated as follows:
where λdaughter and λparent are the decay constants of daughter and parent nuclides, respectively, Ndaughter/Nparent is the amount ratio of the daughter and parent nuclides in the sample, and t is the elapsed time since the separation of the radionuclides. The daughter-to-parent ratio (Ndaughter/Nparent) is often referred to as chronometer, while the elapsed time (t) is called the (model) age of the material. Usually 230Th/234U chronometer is used for age dating of uranium materials. The reason for this is the high chemical dissimilarity between Th and U and the easier measurement of the 230Th in trace level. A major drawback for the Pa measurements is the lack of long-lived Pa isotopes (beside the analyte 231Pa), which can be used as a spike. The often used short-lived 233Pa spike (T1/2 = 26.98 days) is typically milked from 237Np and calibrated against a rock standard (e.g. Table Mountain Latite [8] or measured with gamma spectrometry at fixed geometry to determine the 233Pa concentration [9]. Milking of 233Pa from the parent 237Np is tedious and time consuming, and it cannot be performed before enough 233Pa is produced from the decay of 237Np, which can take weeks to reach the secular equilibrium. As Pa is prone to adsorption, loss of the 233Pa spike cannot be excluded. Another option for milking is to use gamma spectrometry before and after the separation to measure the recovery of 233Pa [10]. The 231Pa analysis can be carried out by alpha spectrometry in a fixed geometry [10] or inductively coupled plasma mass spectrometry (ICP-MS) [8]. It has to be noted that as 231Pa and 233Pa decay with a different mode (alpha and beta decay), thus they cannot be measured simultaneously in a quantitative way by radiometric methods. Hence, mass spectrometry is a viable alternative. With mass spectrometry, the measurement of the short-lived 233Pa is cumbersome, as measurable amount (i.e. relative high activity) is needed for the precise measurement.
The aim of the present study was to develop a simple, but precise and accurate method for the sample preparation and subsequently precise determination of the 231Pa/235U ratio in uranium matrices. In order to minimise the possible loss of Pa during milking and evade it e.g. at the end of the measurement by gamma spectrometry, the 237Np solution in secular equilibrium with its daughter 233Pa was added directly to the sample solution prior to the chemical separation. As more standards are available for 237Np, its measurement is much straightforward than preparing a 233Pa spike solution. The direct spiking does not require the regular milking of 233Pa after the ingrowth from 237Np, so the Pa spike is readily and continuously available. The Pa/Np fractionation was checked with gamma spectrometry. The developed method was applied for four U certified reference materials (CRMs) either with certified (model) age determined from the 230Th/234U ratio or with known production history.
Experimental
Reagents and materials
All labware was thoroughly cleaned before use. Suprapur grade hydrochloric, hydrofluoric and nitric acids (Merck, Darmstadt, Germany) were used for the sample preparation. HNO3 was further purified by sub-boiling distillation (AHF Analysentechnik AG, Germany). For dilutions ultrapure water was used (Elga LabWater, Celle, Germany). A 233U isotopic reference material was used to spike the samples for the uranium concentration measurements by isotope dilution mass spectrometry. The 233U concentration in the spike was calibrated against EC NRM 101 uranium metal by thermal ionization mass spectrometry (TIMS). Uranium CRM U-010 standard reference material (nominally 1% 235U) from National Bureau of Standards (USA) was used to correct for instrumental mass discrimination of the ICP-MS. Isotopic reference material IRMM-185 (certified n(235U)/n(238U) value is (2.00552 ± 0.00060) × 10−2) was used as a quality control sample for the uranium (and protactinium) isotope ratio measurements. The 237Np spike was prepared from certified reference material solution from Cetama (CEA, France). The 237Np solution was transferred to a Teflon vial and kept under weight control. The solution medium was ccHCl/ccHNO3 to avoid precipitation or adsorption. The 237Np concentration was around 900 μg/g, while the n(231Pa)/n(233Pa) amount ratio was 0.0464 ± 0.0013. The resin retention studies were performed using a carrier-free 231Pa solution (PNP10010) from AEA Technology (UK). The activity concentration was 419 Bq/g.
TK-400 (50–100 μm particle size) extraction chromatographic resin was supplied by Triskem International (Bruz, France). For column preparation 1.8 ml resin was used in a polyethylene Bio-Rad holder (diameter: 6 mm), washed and conditioned with 8 ml ccHCl before use. Silica gel (Merck KGaA, 10-40 μm particle size, 0.5 ml bed volume, column diameter: 4 mm) was filled in as slurry in a polyethylene Bio-Rad column holder. The column was washed and conditioned with 3 ml of 4% HNO3. Porous Teflon frits (Reichelt Chemietechnik Heidelberg, Germany) were placed gently on the top of the resins to avoid mixing.
Instrumentation
The Pa, Np and U isotopic analyses were performed using a double-focusing magnetic sector inductively coupled plasma mass spectrometer (ICP-MS) equipped with a single electron multiplier (Element2, Thermo Electron Corp., Bremen, Germany). All measurements were done in low resolution mode (R = 300) using a low-flow micro-concentric nebulizer operated in a self-aspirating mode (flow rate was approximately 50 μL/min) in combination with a quartz Stable Introduction System. Concentrations of isotopes of interest were determined as a function of 231Pa/233Pa and 233U/235U ratios according to the isotope dilution method (ID-MS). The measured amount contents of 231Pa and 235U were used to calculate the model ages according to Eq. (1). The measured isotope ratios obtained by ICP-MS were corrected for instrumental mass bias using linear correction [11]. The 237Np spike concentration was determined by ICP-MS using external calibration and Bi as an internal standard. The U concentrations and isotopic compositions were also measured by thermal ionization mass spectrometry (TIMS) using a Triton instrument (Thermo Scientific, Bremen, Germany) for U isotopics to confirm the ICP-MS results, but they were not used for the evaluation.
Optimization of the Pa and U separation was monitored by high resolution gamma spectrometry (HRGS) using a well-type HPGe detector (GCW 2022 model, Canberra Industries Inc., USA) with approximately 20% relative efficiency and a resolution of < 1.7 keV at 185.6 keV. The gamma counting system consisted of a Canberra model 2022 amplifier and a Canberra model 8075 analog-to-digital converter. The measured spectra were evaluated using Genie 2000 v2.1 software. The measurement times varied between 600 and 5200 s. All gamma spectrometric measurements were performed at fixed geometries (i.e. relative measurements to the original starting material before the separation). The 49.55 keV gamma peak of 238U (emission probability of 0.064%) and 27.4 keV gamma peak of 231Pa (emission probability of 11.1%) were used. Background was measured every day.
In order to measure the 237Np content and Pa/Np ratio, a sample containing 10 ml of 1 mg/ml 237Np stock solution was measured by an extended range HPGe detector with 50% relative efficiency and a resolution of < 1.9 keV at 1.3 MeV using a LabSOCS™ (Laboratory Sourceless Calibration Software) mathematical efficiency calibration software. Spectra were collected for 60,000 s with a DSA-1000 Digital Spectrum Analyzer and evaluated using Genie 2000 v.3.2.1 software. Efficiency calibration was calculated with LabSOCS™. 237Np activity was evaluated from the gamma lines at 143.3 and 151.4 keV; for 233Pa the lines at 300.3, 312.2, 340.8, 398.6 and 415.8 keV were used.
Investigated U samples
For the optimization of the Pa/U separation U3O8 with natural isotopic composition was used [12]. This sample was available in higher quantity and purity. The analysed U reference materials were CRM 125-A (approx. 4% enriched UO2 pellet from New Brunswick Laboratory, USA), IRMM-1000b (approx. 3.6% enriched uranium nitrate from EC Joint Research Centre, Geel, Belgium), U100 (approx. 10% enriched U3O8 from New Brunswick Laboratory, USA) and U630 (approx. 63% enriched U3O8 from New Brunswick Laboratory, USA). The materials have either a certified production date through the 230Th/234U model age (CRM 125-A, IRMM-1000b, U630) or known (archive) production date (U100). 60-80 mg of each of the U materials was dissolved in 8 M HNO3/0.02 M HF on a hot plate at 80 °C for 24 h resulting about 20 mg U/ml solution.
Optimization of Pa separation from U matrix
Two methods were considered for Pa separation: TK-400 extraction chromatography resin [13] and silica gel [8, 10]. Other options, like ion exchange chromatography or extraction chromatography with other resins were excluded, either because of the low Pa recovery or ineffective separation from Np and U [14]. The two methods were tested using natural U3O8 and 231Pa tracer and the measurements were performed by gamma spectrometry. The fractions from the successive elution were collected in 1-ml portions to calculate the U separation factor (defined as the quotient of the Pa/U ratio in the initial material and after the chemical separation) and Pa recovery. HF was removed by adding 20 μl of saturated H3BO3 and a few drops of HClO4. Before loading the solutions in the columns, they were converted to ccHCl (TK-400) or 4% HNO3 (silica gel). The final volume of the test sample for loading was 5 ml (TK-400) or 1 ml (silica gel), with the U amount of 1.5 mg for both cases. For TK-400 test the wash was 6 × 1 ml of ccHCl, and the Pa strip (elution) was completed using 6 × 1 ml of 1 M HCl. For silica gel, as the capacity of the resin is much higher due to the small particle size, after wash with 3 × 1 ml of 4% HNO3, Pa was eluted with 3 × 1 ml of 4% HNO3/0.02 M HF. The elution profiles of the two resins are shown in Fig. 1.

Elution profile of U and Pa using different resins
For TK-400 multiple wash steps were necessary to reduce the U amount in the sample. Collecting the first two Pa strips, a U/Pa separation factor of 1300 could be achieved with a Pa recovery of 95%. When using the silica gel, much higher separation of U could be achieved (separation factor was 1.0 × 106) with a recovery of 96% using only 1 ml strip solution. As has been shown by other studies, Np behaves similarly to U in the separation scheme [8, 10].
Due to the much higher separation factor, the one-step silica gel column was chosen for the following separations. A rapid separation is important for the short-lived 233Pa. Another advantage is that use of ccHCl can be omitted, which can cause corrosion in the glove-box. Possible clogging of the sample introduction system during the ICP-MS measurement due to the SiO2 particles from the silica gel resin (which was the major concern) was avoided by adding 0.4 ml of 4% HNO3/0.02 M HF to the eluted Pa fraction.
231Pa/235U ratio measurement of the U samples
Aliquots of the dissolved U CRM samples were transferred gravimetrically to Teflon containers. About 20 mg U was used for each measurement. 300 μl of 237Np spike was added gravimetrically to the samples corresponding to about 270 μg 237Np and 10 pg 233Pa. 25 μl of saturated H3BO3 and 30 μl of HClO4 were added to the samples to remove the HF. The samples were evaporated to almost complete dryness. 200 μl ccHNO3 was added to the samples together with 25 μl H3BO3 and 30 μl of HClO4. The samples were evaporated again. Overall, this step was repeated three times. Then the samples were taken up to 1 ml of 4% HNO3. The samples were loaded on the pre-conditioned silica gel column. After loading the samples, the columns were washed with 4 × 1 ml 4% HNO3, and Pa was eluted with 2 × 0.8 ml 4% HNO3/0.02 M HF in a polyethylene vial. The ICP-MS measurement was performed soon after the final wash step and Pa elution in order to minimize the 233U ingrowth from the 233Pa decay. Times of loading, elution and measurement were recorded (see Supplementary Information). Due to the short half-life of 233Pa (decay of 233Pa is about 2.5% in 24 h) the time span between the separation and measurement has to be recorded and taken into account. The time span was defined as the difference of the sample loading and the ICP-MS measurement. As this span is difficult to measure exactly, a 0.5 h uncertainty was assigned to the length of separation. All samples were measured in duplicate referring to #1 and #2.
Data evaluation
For the age calculations nuclear data from the Decay Data Evaluation Project were used [15]. The 231Pa and 235U half-lives are 32,670 ± 260 years and 704 ± 1 × 106 years (k = 1), respectively. The overall uncertainties were calculated according to ISO/BIPM guide and taking into account the uncertainty of the weight measurements, spike concentrations, measured isotope ratios, nuclear data and elapsed time between the separation and measurement [16]. The given uncertainties are expanded uncertainties with a coverage factor of k = 2 if not indicated otherwise. The Pa chemical recovery and U separation factor calculations were carried out by Excel®, while for the age calculations commercially available software, GUM Workbench was used [17]. The schematic of the age measurement is shown in Fig. 2.

A schematic of the age determination from the 231Pa/235U ratio
Results and discussion
237Np spike measurement
The 237Np spike, which is in secular equilibrium with 233Pa, was measured by HRGS and by ICP-MS for the 237Np concentration. The HRGS and ICP-MS resulted in 903 ± 45 μg/g (k = 1) and 890 ± 9 μg/g (k = 1), respectively. The determined values based on different principles (activity vs. mass) resulted in comparable results (Fig. 3). A combined value was obtained from the different results by taking the mean weighted by the inverse of the variances [15]. The combined value of 896 ± 10 μg/g (k = 1) was used for the chronometric studies.

Measurement results of the 237Np spike concentration
It has to be noted that 233Pa amount can also be measured with gamma spectrometry, not only the 237Np amount in the sample. However, in order to compare the 237Np values obtained by HRGS and ICP-MS result the 233Pa result was not used. With gamma spectrometry the possible adsorption of Pa on the inner surface of the vial was checked: the Pa/Np ratio was 1.00 ± 0.03 with the standing vial, while turning it upside down the ratio was 0.96 ± 0.06. If Pa would adsorb on the surface of the vial, the Pa/Np ratio would have been much lower in the latter case as the Pa efficiency is lower due to the higher distance from the detector. It was concluded that Pa was in the liquid phase together with Np, and no fractionation had occurred between the radionuclides, i.e. no adsorption had taken place on the vial surfaces.
231Pa/235U model production date results
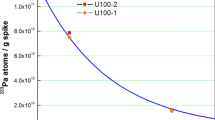
A typical ICP-MS spectrum is shown in Fig. 4. Even though there was only a single separation, U and Np were well separated from the Pa fraction. The 231Pa/235U mass ratios together with the 231Pa/235U model ages and production dates are summarized in Table 1. The measured 231Pa and 235U concentrations used for the model age calculation are collected in the Supplementary Information. The 235U values measured by isotope dilution TIMS were used to check the ICP-MS results, but they were not used for the model age calculation.

Typical ICP-MS spectrum (U100 #1). The shown mass regions are the selected masses for measurement (m/z of 231Pa, 233Pa, 234U and 237Np). Major U isotopes were not measured due to the possible high signal
The replicate results agree well with each other. The age values have a relative expanded uncertainty of 2.7–3.9%, which is determined mainly by the low abundant 231Pa measurement: about 90–95% of the total uncertainty contribution derives from the 231Pa analysis. Out of the 231Pa measurement uncertainty the major components are 231Pa/233Pa ratio measurements in the blend by ICP-MS and 237Np (233Pa) spike concentration. Other parameters, like weight measurement, nuclear data, length of separations or other ratio measurements, correspond to less than 10% of the to the total uncertainty.
The measured values agree well with the reported values [8]. For three materials, CRM 125-A, IRMM-1000 and U630, the measured 231Pa/235U model ages are shown together with the certified production dates (Fig. 5). As the obtained 231Pa/235U ages agree with the ages obtained from the 230Th/234U ratio, it indicates that at the time of production of these materials Th and Pa had been quantitatively removed from uranium. For IRMM-1000 both obtained values are slightly lower than the certified age. Although this is not significant, it was also observed during the REIMEP-22 inter-laboratory comparison [18]. For U100 the measured 231Pa/235U production dates are in agreement with the purification date of 8 January, 1959 [8].

231Pa/235U model production date of the measured U samples. In a, b, d the certified production dates by the 230Th/234U ratio are also given
Conclusions
An improved method was developed for the chemical separation of Pa and determination of the production date of uranium materials based on the 231Pa/235U ratio measurement. The 233Pa spike necessary for the quantification of the 231Pa concentration was obtained by adding 237Np solution directly to the sample, where the 233Pa was in secular equilibrium with its parent nuclide. Therefore, there is no need to separate beforehand 233Pa from 237Np, and the spike is continuously available without the need of milking. Omitting the milking step the possibility of Pa loss through adsorption is reduced significantly. The possible fractionation between Np and Pa as well as the potential adsorption of the spike was checked by gamma spectrometry. The proposed one-step separation and the measurement method are faster and lower uncertainty can be achieved than with alpha spectrometry and it is applicable even for young low-enriched uranium samples. The method can be applied for uranium found out of regulatory control (i.e. in nuclear security) and for safeguards samples to complement the 230Th/234U model age, i.e. to check if concurrent ages are obtained.
References
Mayer K, Wallenius M, Varga Z (2013) Nuclear forensic science: correlating measurable material parameters to the history of nuclear material. Chem Rev 113(2):884–900
Kristo MJ, Tumey SJ (2013) The state of nuclear forensics. Nucl Instrum Methods Phys Res Sect B 294:656–661
Wallenius M, Mayer K, Ray I (2006) Nuclear forensic investigations: two case studies. Forensic Sci Int 156:55–62
Han SH, Varga Z, Krajkó J, Wallenius M, Song K, Mayer K (2013) Measurement of the sulphur isotope ratio (34S/32S) in uranium ore concentrates (yellow cakes) for origin assessment. J Anal At Spectrom 28(12):1919–1925
Wallenius M, Morgenstern A, Apostolidis C, Mayer K (2002) Determination of the age of highly enriched uranium. Anal Bioanal Chem 374:379–384
Williams RW, Gaffney AM (2011) 230Th-234U model ages of some uranium standard reference materials. Proc Radiochim Acta 1:31–35
Varga Z, Surányi G (2007) Production date determination of uranium-oxide materials by inductively coupled plasma mass spectrometry. Anal Chim Acta 599:16–23
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U-231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom 28(5):666–674
Mendes M, Aupiais J, Jutier C, Pointurier F (2013) Determination of weight distribution ratios of Pa(V) and Np(V) with some extraction chromatography resins and the AG1-X8 resin. Anal Chim Acta 780:110–116
Morgenstern A, Apostolidis C, Mayer K (2002) Age determination of highly enriched uranium: separation and analysis of 231Pa. Anal Chem 74:5513–5516
Heumann KG, Gallus SM, Rädlinger G, Vogl J (1998) Precision and accuracy in isotope ratio measurements by plasma source mass spectrometry. J Anal At Spectrom 13:1001–1008
Varga Z, Krajkó J, Peńkin M, Novák M, Eke Z, Wallenius M, Mayer K (2017) Identification of uranium signatures relevant for nuclear safeguards and forensics. J Radioanal Nucl Chem 312(3):639–654
Knight AW, Nelson AW, Eitrheim ES, Forbes TZ, Schultz MK (2016) A chromatographic separation of neptunium and protactinium using 1-octanol impregnated onto a solid phase support. J Radioanal Nucl Chem 307(1):59–67
Jerome SM, Collins SM, Happel S, Ivanov P, Russell BC (2018) Isolation and purification of protactinium-231. Appl Radiat Isot 134:18–22
DDEP Monographie BIPM-5—”Table of Radionuclides” (2015) http://www.nucleide.org/DDEP.htm. Accessed 1 Sept 2018
Joint Committee for Guides in Metrology Evaluation of measurement data—guide to the expression of uncertainty in measurement. JCGM 100:2008 (2008)
GUM Workbench Pro, Version 2.3.6.127 (2009) Metrodata GmbH, Weil am Rhein, Germany
Venchiarutti C, Varga Z, Richter S, Jakopič R, Mayer K, Aregbe Y (2015) REIMEP-22 inter-laboratory comparison: “U Age Dating—determination of the production date of a uranium certified test sample”. Radiochim Acta 103(12):825–834
Acknowledgements
The EC JRC-Karlsruhe Analytical Service is kindly acknowledged for their valuable assistance.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Varga, Z., Nicholl, A., Hrnecek, E. et al. Measurement of the 231Pa/235U ratio for the age determination of uranium materials. J Radioanal Nucl Chem 318, 1565–1571 (2018). https://doi.org/10.1007/s10967-018-6247-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-018-6247-9