
Abstract
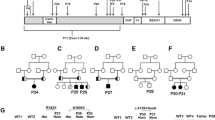
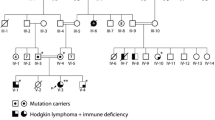
Pathogenic variants in LRBA, encoding the LPS Responsive Beige-Like Anchor (LRBA) protein, are responsible for recessive, early-onset hypogammaglobulinemia, severe multi-organ autoimmunity, and lymphoproliferation, with increased risk for malignancy. LRBA deficiency has a wide clinical spectrum with variable age of onset and disease severity. Three apparently unrelated patients with LRBA deficiency, of Georgian Jewish descent, were homozygous for LRBA c.6640C > T, p.R2214*, leading to a stop upstream of the LRBA BEACH domain. Despite carrying the same LRBA genotype, the three patients differed in clinical course: the first patient was asymptomatic until age 25 years; the second presented with failure to thrive at age 3 months; and the third presented at age 7 years with immune cytopenias and severe infections. Two of the patients developed malignancies: the first patient was diagnosed with recurrent Hodgkin’s disease at age 36 years, and the second patient developed aggressive gastric cancer at age 15 years. Among Georgian Jews, the carrier frequency of the LRBA p.R2214* allele was 1.6% (4 of 236 Georgian Jewish controls). The allele was absent from other populations. Haplotype analysis showed a shared origin of the mutation. These three patients revealed a pathogenic LRBA founder allele in the Georgian Jewish population, support the diverse and complex clinical spectrum of LRBA deficiency, and support the possibility that LRBA deficiency predisposes to malignancy.






Similar content being viewed by others

Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24–64. https://doi.org/10.1007/s10875-019-00737-x.
Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. The ever-increasing array of novel inborn errors of immunity: an interim update by the IUIS committee. J Clin Immunol. 2021;41(3):666–79.
Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, members of the CEREDIH French PID study group. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol. 2017;140(5):1388-1393.e8.
Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90(6):986–1001.
Charbonnier LM, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT, et al. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol. 2015;135(1):217–27. https://doi.org/10.1016/j.jaci.2014.10.019.
Revel-Vilk S, Fischer U, Keller B, Nabhani S, Gamez-Diaz L, Rensing-Ehl A, et al. Autoimmune lymphoproliferative syndrome-like disease in patients with LRBA mutation. Clin Immunol. 2015;159(1):84–92.
Gamez-Diaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. 2016;137(1):223–30.
Cagdas D, Halacli SO, Tan C, Lo B, Cetinkaya PG, Esenboga S, et al. A spectrum of clinical findings from ALPS to CVID: several novel LRBA defects. J Clin Immunol. 2019;39(7):726–38.
Meshaal S, El Hawary R, Adel R, AbdElaziz D, Erfan A, Lotfy S, et al. Clinical phenotypes and immunological characteristics of 18 Egyptian LRBA deficiency patients. J Clin Immunol. 2020;40(6):820–32.
Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol. 2016;36(1):33–45.
Soler-Palacin P, Garcia-Prat M, Martin-Nalda A, Franco-Jarava C, Riviere JG, Plaja A, et al. LRBA deficiency in a patient with a novel homozygous mutation due to chromosome 4 segmental uniparental isodisomy. Front Immunol. 2018;16(9):2397.
Bratanic N, Kovac J, Pohar K, TrebusakPodkrajsek K, Ihan A, Battelino T, et al. Multifocal gastric adenocarcinoma in a patient with LRBA deficiency. Orphanet J Rare Dis. 2017;12(1):131–017.
Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349(6246):436–40.
Seidel MG, Bohm K, Dogu F, Worth A, Thrasher A, Florkin B, et al. Treatment of severe forms of LPS-responsive beige-like anchor protein deficiency with allogeneic hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2018;141(2):770-775.e1.
Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol. 2020;145(5):1452–1463. https://doi.org/10.1016/j.jaci.2019.12.896
Baxter SK, Walsh T, Casadei S, Eckert MM, Allenspach EJ, Hagin D, et al. Molecular diagnosis of childhood immune dysregulation, polyendocrinopathy, and enteropathy, and implications for clinical management. J Allergy Clin Immunol. 2022;149(1):327–39.
Warnatz K, Wehr C, Drager R, Schmidt S, Eibel H, Schlesier M, et al. Expansion of CD19(hi)CD21(lo/neg) B cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology. 2002;206(5):502–13.
Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115(24):5026–36.
Keller B, Strohmeier V, Harder I, Unger S, Payne KJ, Andrieux G, et al. The expansion of human T-bet(high)CD21(low) B cells is T cell dependent. Sci Immunol. 2021;6(64):eabh0891. https://doi.org/10.1126/sciimmunol.abh0891.
Hou TZ, Verma N, Wanders J, Kennedy A, Soskic B, Janman D, et al. Identifying functional defects in patients with immune dysregulation due to LRBA and CTLA-4 mutations. Blood. 2017;129(11):1458–68.
Alexander Z. Immigration to Israel from the USSR. : Faculty of Law, Tel Aviv University; 1977.
Thomas MG, Weale ME, Jones AL, Richards M, Smith A, Redhead N, et al. Founding mothers of Jewish communities: geographically separated Jewish groups were independently founded by very few female ancestors. Am J Hum Genet. 2002;70(6):1411–20.
Behar DM, Metspalu E, Kivisild T, Rosset S, Tzur S, Hadid Y, et al. Counting the founders: the matrilineal genetic ancestry of the Jewish Diaspora. PLoS One. 2008;3(4):e2062.
Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370(10):921–31.
Acknowledgements
We would like to thank our patients and their families for their support and collaboration. In addition, we thank Prof. Ephrat Levy-Lahad (Director of the Medical Genetics Institute at the Shaare Zedek Medical Center, Jerusalem, Israel) for sharing DNA samples of Georgian Jewish donors.
Funding
D.H. is funded by the Alrov Foundation and the Joint Tel-Aviv Sourasky Medical Center and The Weizmann Institute of Science Research Grant. S.K.B., T.W., and M.C.K. were funded by National Institutes of Health grant R35 CA197458.
Author information
Authors and Affiliations
Contributions
TF, SB, and DH: study conception and design, acquisition of data, analysis and interpretation of data, and manuscript preparation. TW, YA, RS, and PR: acquisition of data, analysis and interpretation of data, and manuscript preparation. HG, JK, MAG, SB, NS, and RR: provided critical clinical care and data. MCK and TRT: analysis and interpretation of data and manuscript preparation.
Corresponding author
Ethics declarations
Ethics Approval
Appropriate ethics committee approvals were obtained at the Tel-Aviv Sourasky Medical Center (Tel-Aviv, Israel) and the Shaare Zedek Medical Center (Jerusalem, Israel).
Consent to Participate
Informed consent was obtained from all individual participants included in the study, with the exeption of patient 2 who sadly died long before the study was designed. Genetic analysis of patient 2’s sample was approved by local IRB.
Consent for Publication
The manuscript does not contain any individual person’s identifying details, images, or videos.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Freund, T., Baxter, S.K., Walsh, T. et al. Clinically Complex LRBA Deficiency Due to a Founder Allele in the Georgian Jewish Population. J Clin Immunol 43, 151–164 (2023). https://doi.org/10.1007/s10875-022-01358-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-022-01358-7