
Abstract
Introduction
Autosomal recessively inherited lipopolysaccharide-responsive beige-like anchor (LRBA) protein deficiency was shown to be responsible for different types of inborn errors of immunity, such as common variable immunodeficiency (CVID) and autoimmune lymphoproliferative syndrome (ALPS). The aim of this study was to compare patients with LRBA-related ALPS and LRBA-related CVID, to describe their clinical and laboratory phenotypes, and to prepare an algorithm for their diagnosis and management.
Methods
Fifteen LRBA-deficient patients were identified among 31 CVID and 14 possible ALPS patients with Western blotting (WB), primary immunodeficiency disease (PIDD) gene, next-generation panel screening (NGS), and whole exome sequencing (WES).
Results
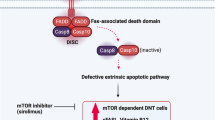
The median age on admission and age of diagnosis were 7 years (0.3–16.5) and 11 years (5–44), respectively. Splenomegaly was seen in 93.3% (14/15) of the patients on admission. Splenectomy was performed to 1/5. Recurrent upper respiratory tract infections (93.3% (14/15)), autoimmune cytopenia (80% (12/15)), chronic diarrhea (53.3% (8/15)), lower respiratory tract infections (53.3% (8/15)), lymphoma (26.6% (4/15)), Evans syndrome (26.6% (4/15)), and autoimmune thyroiditis (20% (3/15)) were common clinical findings and diseases. Lymphopenia (5/15), intermittant neutropenia (4/15), eosinophilia (4/15), and progressive hypogammaglobulinemia are recorded in given number of patients. Double negative T cells (TCRαβ+CD4−CD8−) were increased in 80% (8/10) of the patients. B cell percentage/numbers were low in 60% (9/15) of the patients on admission. Decreased switched memory B cells, decreased naive and recent thymic emigrant (RTE) Thelper (Th) cells, markedly increased effector memory/effector memory RA+ (TEMRA) Th were documented. Large PD1+ population, increased memory, and enlarged follicular helper T cell population in the CD4+ T cell compartment was seen in one of the patients. Most of the deleterious missense mutations were located in the DUF1088 and BEACH domains. Interestingly, one of the two siblings with the same homozygous LRBA defect did not have any clinical symptom. Hematopoietic stem cell transplantation (HSCT) was performed to 7/15 (46.6%) of the patients. Transplanted patients are alive and well after a median of 2 years (1–3). In total, one patient died from sepsis during adulthood before HSCT.
Conclusion
Patients with LRBA deficiency may initially be diagnosed as CVID or ALPS in the clinical practice. Progressive decrease in B cells as well as IgG in ALPS-like patients and addition of IBD symptoms in the follow-up should raise the suspicion for LRBA deficiency. Decreased switched memory B cells, decreased naive and recent thymic emigrant (RTE) Th cells, and markedly increased effector memory/effector memory RA+ Th cells (TEMRA Th) cells are important for the diagnosis of the patients in addition to clinical features. Analysis of protein by either WB or flow cytometry is required when the clinicians come across especially with missense LRBA variants of uncertain significance. High rate of malignancy shows the regulatory T cell’s important role of immune surveillance. HSCT is curative and succesful in patients with HLA-matched family donor.




Similar content being viewed by others


References
Alangari A, et al. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol. 2012;130(2):481–488. e2.
Lopez-Herrera G, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90(6):986–1001.
Bogaert DJ, et al. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. 2016;53(9):575–90.
Revel-Vilk S, et al. Autoimmune lymphoproliferative syndrome-like disease in patients with LRBA mutation. Clin Immunol. 2015;159(1):84–92.
Lo B, et al. CHAI and LATAIE: new genetic diseases of CTLA-4 checkpoint insufficiency. Blood. 2016;128:1037–42. https://doi.org/10.1182/blood-2016-04-712612.
Serwas NK, et al. Atypical manifestation of LRBA deficiency with predominant IBD-like phenotype. Inflamm Bowel Dis. 2014;21(1):40–7.
Burns SO, et al. LRBA gene deletion in a patient presenting with autoimmunity without hypogammaglobulinemia. J Allergy Clin Immunol. 2012;130(6):1428–32.
Alroqi FJ, et al. Exaggerated follicular helper T-cell responses in patients with LRBA deficiency caused by failure of CTLA4-mediated regulation. J Allergy Clin Immunol. 2018;141(3):1050–1059.e10.
Gámez-Díaz L, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. 2016;137(1):223–30.
Oliveira JB, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome: report from the 2009 NIH International Workshop. Blood. 2010;116(14):e35–40. https://doi.org/10.1182/blood-2010-04-280347.
Lo B, et al. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349(6246):436–40.
Ozen A, et al. CD55 deficiency, early-onset protein-losing enteropathy, and thrombosis. N Engl J Med. 2017;377(1):52–61.
Willmann, K.L., et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun. 2014;5:5360
Al-Mousa H, et al. High incidence of severe combined immunodeficiency disease in Saudi Arabia detected through combined TREC and next generation sequencing of newborn dried blood spots. Front Immunol. 2018;9:782.
Kostel Bal S, et al. Multiple presentations of LRBA deficiency: a single-center experience. J Clin Immunol. 2017;37(8):790–800.
Okur F, et al. Bone marrow transplantation with favorable outcome in three patients with LPS-responsive beige-like anchor (LRBA) deficiency. Clin Immunol (Orlando, Fla). 2019;203:162.
Johnson MB, et al. Recessively inherited LRBA mutations cause autoimmunity presenting as neonatal diabetes. Diabetes. 2017;66(8):2316–22. https://doi.org/10.2337/db17-0040.
Cullinane AR, Schaffer AA, Huizing M. The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic. 2013;14(7):749–66.
Tan Ç, et al. Polymorphisms in FAS and CASP8 genes may contribute to the development of ALPS phenotype: a study in 25 patients with probable ALPS. Turk J Pediatr. 2015;57(2):141.
Lee K-M, et al. Molecular basis of T cell inactivation by CTLA-4. Science. 1998;282(5397):2263–6.
Riaz IB, et al. A systematic review on predisposition to lymphoid (B and T cell) neoplasias in patients with primary immunodeficiencies and immune dysregulatory disorders (inborn errors of immunity). Front Immunol. 2019;10:777.
Alkhairy OK, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol. 2016;36(1):33–45.
Tarbox JA, et al. Elevated double negative T cells in pediatric autoimmunity. J Clin Immunol. 2014;34(5):594–9.
Lévy E, et al. LRBA deficiency with autoimmunity and early onset chronic erosive polyarthritis. Clin Immunol. 2016;168:88–93.
Azizi G, et al. New therapeutic approach by sirolimus for enteropathy treatment in patients with LRBA deficiency. Eur Ann Allergy Clin Immunol. 2017;49(5):235–9.
Jung SW, et al. BK virus-associated nephropathy with hydronephrosis in a patient with AIDS: a case report and literature review. Clin Nephrol. 2016;85(3):173–8.
Azizi G, et al. Polyautoimmunity in patients with LPS-responsive beige-like anchor (LRBA) deficiency. Immunol Investig. 2018;47(5):457–67.
Jogl G, et al. Crystal structure of the BEACH domain reveals an unusual fold and extensive association with a novel PH domain. EMBO J. 2002;21(18):4785–95.
Schubert D, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410.
Seidel MG, et al. Long-term remission after allogeneic hematopoietic stem cell transplantation in LPS-responsive beige-like anchor (LRBA) deficiency. J Allergy Clin Immunol. 2015;135(5):1384–1390. e8.
Seymour B, Miles J, Haeney M. Primary antibody deficiency and diagnostic delay. J Clin Pathol. 2005;58(5):546–7.
Acknowledgments
We are thankful to our patients and families. We appreciate our laboratory staff Ozlem Karapınar, Mehtap Sonmez, Menekse Kocak and our nurses Meliha Erol and Feride Ozkan. This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. We thank Merck and Co. for support.
Funding
This study includes the data of three studies (Project codes: 013 D08103 001-341, 11/19-23, and 16/9087) which are funded by Hacettepe University Coordination Unit for Scientific Research Projects. The study is additionally funded by TUBITAK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The studies were approved by the Institutional Review Board of Hacettepe University.
Conflict of Interest
The authors declare that they have no conflict of interest
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 26.8 KB)
Rights and permissions
About this article

Cite this article
Cagdas, D., Halaçlı, S.O., Tan, Ç. et al. A Spectrum of Clinical Findings from ALPS to CVID: Several Novel LRBA Defects. J Clin Immunol 39, 726–738 (2019). https://doi.org/10.1007/s10875-019-00677-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-019-00677-6