
Abstract
The title compound, Abacavir hemisulfate [Systematic name: {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopentene}-1-methanol hemisulfate, [C14H19N6O+]2 SO4 2−, (I), crystallizes in the monoclinic space group C2 with unit cell parameters a = 18.7498(7), b = 8.3577(5), c = 13.2563(5), Å, β = 130.575(3)°, Z = 2. In the asymmetric unit, the compound crystallizes with two C14H19N6O+ cations and one SO4 2− anion connected by strong intermolecular N–H···O hydrogen bond interactions between two of these C14H19N6O+ cations. Additional strong O–H···O hydrogen bond intermolecular interactions are observed between the C14H19N6O+ and SO4 2−, cation–anion, units, respectively. The [C14H19N6O+]2 SO4 2− units link themselves into an infinite O–H···O–H···O–H one dimensional chain diagonally along the [101] plane of the unit cell. The dihedral angle between the mean planes of the 5-membered imidazole ring fused to the 6-membered pyrimidine ring is 2.6(8)° making it a nonplanar purin-9-yl ring. The dihedral angle between the mean planes of the imidazole and pyrimidine rings with those of the cyclopropane and cyclopentene rings are 63.3(7)°, 61.2(2)° and 63.2(2)°, 63.87°, respectively. Intermolecular O–H···O and N–H···O hydrogen bond interactions influence the cyclopropane twist angle and therefore play a role in stabilizing crystal packing in the unit cell. The geometries of the C14H19N6O+, SO4 2−, cation–anion pair, were optimized means of density functional theory (DFT) molecular orbital theoretical calculation at the B3-LYP/6-311 + G(d,p) level and compared to the crystallographic results providing support for these observations.
Graphical Abstract
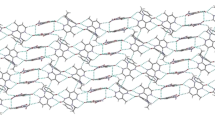
Crystal structure of Abacavir hemisulfate is reported and its geometric and packing parameters described and compared to a DFT computational calculation.





Similar content being viewed by others


Notes
In this approach a density synthesis is calculated around the circle which represents the locus of possible hydrogen positions (for a fixed X–H distance and Y–X–H angle). The maximum electron density is then taken as the starting position for the hydrogen atom. In subsequent refinement cycles (and in further least-squares jobs) the hydrogen is re-idealized at the start of each cycle, but the current torsion angle is retained; the torsion angles are allowed to refine whilst keeping the X–H distance and Y–X–H angle fixed.
References
Hervey PS, Perry CM (2000) Drugs 60:447–479
Clay PG (2002) Clin Ther 24:1502–1514
Sheldrick GM (2008) Acta Cryst A64:112–122
Bruker (2006) SHELXTL. Bruker AXS Inc, Madison
Allen FH, Kennard O, Watson DG, Brammer L, Orpen AG, Taylor RJ (1987) Chem Soc Perkin Trans 2:S1–S19
Monger G, Varlashkin P (2005) Powder Diffract 20:241–245
Sheldrick GM (1997) SHELXL-97 reference manual. University of Gottingen
Bernstein J, Davis RE, Shimoni L, Chang NL (1995) Angew Chem Int Ed 34:1555–1573. doi:10.1002/anie.199515551
Schmidt JR, Polik WF (2007) WebMO Pro, version 8.0.010e; WebMO, LLC, Holland, MI, USA. Available from http://www.webmo.net
Frisch MJ et al (2004) Gaussian 03, Revision C01. Wallingford, CT
Becke AD (1988) Phys Rev A38:3098–3100
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B37:785–789
Hehre WJ, Random L, Schleyer PvR, Pople JA (1986) Ab Initio Molecular Orbital Theory. Wiley, New York
Acknowledgments
HSY thanks the University of Mysore for use of their research facilities. R. J. B acknowledges the NSF MRI program (grant no. CHE-0619278) for funds to purchase the X-ray diffractometer.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jasinski, J.P., Butcher, R.J., Yathirajan, H.S. et al. Crystal Structure of Abacavir Hemisulfate: A Nucleoside Analog Reverse Transcriptase Inhibitor. J Chem Crystallogr 39, 864–869 (2009). https://doi.org/10.1007/s10870-009-9577-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-009-9577-1