
Abstract
Data integration in structural biology has become a paradigm for the characterization of biomolecular systems, and it is now accepted that combining different techniques can fill the gaps in each other’s blind spots. In this frame, one of the combinations, which we have implemented in REFMAC-NMR, is residual dipolar couplings from NMR together with experimental data from X-ray diffraction. The first are exquisitely sensitive to the local details but does not give any information about overall shape, whereas the latter encodes more the information about the overall shape but at the same time tends to miss the local details even at the highest resolutions. Once crystals are obtained, it is often rather easy to obtain a complete X-ray dataset, however it is time-consuming to obtain an exhaustive NMR dataset. Here, we discuss the effect of including a-priori knowledge on the properties of the system to reduce the number of experimental data needed to obtain a more complete picture. We thus introduce a set of new features of REFMAC-NMR that allow for improved handling of RDC data for multidomain proteins and multisubunit biomolecular complexes, and encompasses the use of pseudo-contact shifts as an additional source of NMR-based information. The new feature may either help in improving the refinement, or assist in spotting differences between the crystal and the solution data. We show three different examples where NMR and X-ray data can be reconciled to a unique structural model without invoking mobility.
Similar content being viewed by others


Introduction
Integrated Structural Biology tries to merge the results from different experimental techniques, making the most of the information encoded in each (Ward et al. 2013; van den Bedem and Fraser 2015; Carlon et al. 2016b; Schlundt et al. 2017). By and large, the two most common experimental techniques for biomolecular structure determination are X-ray diffraction (no. pdb entries retrieved on October 2, 2018: 129581/144682, i.e. 89.6%) and solution NMR spectroscopy (no. pdb entries: 12303/144682, i.e. 8.5%), and single particle cryo electron microscopy (no. pdb entries: 2434/144682, i.e. 1.7%) is progressing rapidly. X-ray crystallographic diffraction (and cryo-EM) data give information that progress from the overall shape of the molecule up to individual atom positions as the resolution increases; on the contrary, NMR data mostly progress from short-range inter-atom distances and bond orientations to the overall shape of the molecule with increasing number and quality of restraints. The combination of these techniques thus yields valuable information throughout the whole range of distances, even in the presence of suboptimal X-ray and/or NMR data.
It is often the case that the X-ray structures and the NMR data are not in perfect agreement with each other. Quite often, these inconsistencies are interpreted as significant and subsequently reconciled by considering structural rearrangements on passing from solid state to solution and/or invoking mobility of different extents and on different timescales (Chou et al. 2001; Bertini et al. 2004, 2009; Volkov et al. 2006; Tang et al. 2007; Lange et al. 2008; Cerofolini et al. 2013; Fenwick et al. 2014; Carlon et al. 2016a, b; Andrałojć et al. 2017). However, before following this path, one should consider first if the X-ray data has sufficient information to position atoms and bonds that are observed by NMR, because, if placed with a low accuracy, inconsistencies may lose significance (structural noise) (Zweckstetter and Bax 2002).
X-ray data, NMR-derived distance restraints and backbone dihedral angles were previously used for a joint refinement of protein structure (Shaanan et al. 1992). It was noticed that crystal models frequently present a large number of NOE distance violations, as well as solution models obtained by NMR poorly fit the X-ray data: these discrepancies may either be due to differences in the molecular structure between solution and solid state, or to the different but complementary information contained in these two types of data. The fact that X-ray and NMR data can be combined to produce models that are compatible with both sets of data, in the sense that (i) the crystallographic R-factor and the free R-factor are the same to those calculated with X-ray data alone, and that (ii) the NMR restraint violations are minimal, indicates that the discrepancies between solution and crystal structures may be apparent rather than real, and that a joint refinement against all data may provide a more reliable model than those obtained using one of the techniques alone. On the other hand, the violating restraints in the joint refinement may provide an indication of the regions where real differences occur.
Joint refinements against X-ray and NMR data were previously performed using distance restraints and backbone dihedral angles as NMR restraints, through the programs CNS and X-PLOR (Brunger et al. 1987). The calculations indicated that the two sets of data are consistent for a number of studied proteins (interleukin-1 β (Shaanan et al. 1992), bovine pancreatic trypsin inhibitor (Schiffer et al. 1994), p53 (Miller et al. 1996), HU (Raves et al. 2001), the integral membrane protein complex DsbB-DsbA (Tang et al. 2011), mostly improving the geometry of the model in terms of Ramachandran plot with respect to the structure calculated without NMR data. In some cases, the joint refinement clearly provided more accurate models, for instance in the presence of regions poorly determined by X-ray data alone, due to packing disorder within the crystal [L9 protein (Hoffman et al. 1996)], or with low- to medium-resolution diffraction data [maltose binding protein-L30e fusion protein in complex with RNA (Chao and Williamson 2004)].
Paramagnetic data such as pseudocontact shifts (PCSs) and residual dipolar couplings (RDCs) carry long-range information on the relative position—or internuclear vector orientation—of the NMR-active nuclei in a protein with respect to a common frame, and thus result very useful to validate and refine the global fold of a protein in solution when crystallographic data are available.
RDCs that originate from partial alignment due to either interaction with an external alignment medium (Tolman et al. 1995) or from the presence of a paramagnetic centre (Banci et al. 1998b), are extremely sensitive reporters of the angles between internuclear vectors and the principal axis frame of the alignment tensor. When RDCs are obtained because of the presence of a paramagnetic centre (Bertini et al. 2002a; Volkov et al. 2006; Clore 2011; Koehler and Meiler 2011; Knight et al. 2013; Hass and Ubbink 2014; Nitsche and Otting 2017; Ravera et al. 2017), PCSs also arise and provide information about the positions of nuclei in the Cartesian space defined by the principal axis frame of the magnetic susceptibility tensor associated to the paramagnetic centre (Kurland and McGarvey 1970; Banci et al. 1996; Bertini et al. 2002a, b). Since the alignment is caused by the anisotropy of the magnetic susceptibility, the two experimental datasets are characterized by the same tensor, and PCSs can be used to strengthen the estimate of the tensor to be used for fitting the RDCs (Bertini et al. 2004, 2009).
PCSs and RDCs contain structural information that has proved very helpful for solving protein structures (Gochin and Roder 1995; Banci et al. 1996, 1998b; Bertini et al. 2001; Gaponenko et al. 2004; Diaz-Moreno et al. 2005; Jensen et al. 2006; Schmitz et al. 2012), and they have therefore been included as structural restraints in the most commonly used programs for protein structure determination from NMR data (Banci et al. 1998a, b, 2004; Schwieters et al. 2003; Schmitz and Bonvin 2011; Schmitz et al. 2012).
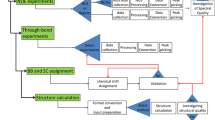
We have included PCSs and RDCs as structural restraints in the Macromolecular Crystal Structure refinement program—REFMAC5 (Murshudov et al. 1997, 2011) available from CCP4 (Winn et al. 2011). This program uses the maximum-likelihood technique to optimize the fit of atomic model parameters into X-ray crystallographic data. The agreement with X-ray data is monitored through the R-factor and the free R-factor, and agreement with the NMR data is monitored through the Q-factor (Cornilescu et al. 1998). As it is common when using X-ray data for structure refinement, some deviations from the ideal geometry of covalent bonding are allowed, and, for this purpose, appropriate weights of the geometric restraints relative to the NMR and X-ray restraints must be selected. If large deviations from ideal geometry are allowed, full compatibility of crystal and NMR data can be achieved. Our strategy for the joint refinement was based on fixing the weights of the geometric restraints to the values providing free R-factor and geometry parameters close to those obtained without including the NMR data. In summary, the applied protocol consists of two steps:
-
1.
standard refinement with REFMAC5 performed using only X-ray data. The final values obtained for R-factor, free R-factor, chemical bonds violations (RMS BondLength, BondAngle and ChirVolume) are taken as reference values.
-
2.
PCS and/or RDC data are added, together with the geometrical restraints. The weight of PCS, RDC and geometrical restraints are changed until the final values of R-factor, free R-factor, and chemical bonds violations are comparable (or better) than the reference values, and PCSs and/or RDCs are fitted as best as possible. This step is repeated until satisfactory values are reached (Q factor < 0.20), or until results show that X-ray and NMR data are in disagreement.
This approach was previously used to show that single refined structures can be calculated using simultaneously X-ray and NMR data collected for the catalytic domain of the protein matrix metalloproteinase 1, for the protein ubiquitin and for the third IgG-binding domain of protein G, thus indicating that no appreciable structural changes occur when passing from solution to solid state; the backbone RMSD between the structures calculated with and without the NMR data is below 0.03 Å. On the contrary, in the case of the N-terminal domain of calmodulin, the joint refinement does not produce any atomic model in agreement with both X–ray and NMR data, indicating that there are some structural differences between the solution and solid state protein (Rinaldelli et al. 2014).
Biomolecules often comprise several domains and subunits, each potentially experiencing an intrinsic variability, and RDCs and PCSs can provide a precious contribution to the characterization of the interdomain/intersubunit arrangement (Valafar and Prestegard 2004; Pintacuda et al. 2007; Bertini et al. 2009, 2010; Simon et al. 2010; Berlin et al. 2010, 2013; Schmitz and Bonvin 2011; Fragai et al. 2013; Cerofolini et al. 2013; Russo et al. 2013; Rinaldelli et al. 2015). In fact, if the complex is composed of domains/proteins with known crystal structures, which do not change in solution upon complex formation, PCSs and RDCs can be very effective in determining their relative position and orientation, once multiple paramagnetic metal ions have been alternatively coordinated to one protein or attached through a rigid binding tag. The process is rather simple. First the RDCs (and PCSs) of each domain/subunit are calculated from an initial model such as the X-ray structure. If a domain or subunit is rigid all the RDCs (and PCSs) referring to atoms belonging to it should fit to a single tensor. If not, one must conclude that the domain is experiencing some static or dynamic rearrangement (Andrałojć et al. 2015; Carlon et al. 2016a).
The second step requires the comparison of the tensors calculated for each domain. If the spatial arrangement of the domains/subunits is the same between the crystal and the solution, all the tensors from each domain will have the same magnitude. If, on the other hand, the tensors have the same magnitude but different orientations, one can conclude that a static rigid rearrangement has occurred upon crystallization (Carlon et al. 2016a, b). If both magnitude and orientation differ, a dynamic process must be present.
The situation described above may not be fulfilled in the presence of experimental uncertainty and in the case where there is not sufficient data to correctly calculate both the tensor and the atomic coordinates. Therefore, the best-fit values of back-calculated tensors may appear more different than needed to be consistent with the experimental data. In this case, one can decide to impose a constraint in the refinement and check whether the agreement between the experimental and calculated NMR data improves or becomes worse. In the first case, the constraint will have helped in improving the refinement, in the latter case it will have assisted in spotting differences between the crystal and the solution data.
We have thus modified the REFMAC-NMR program for either constraining the alignment tensors to a unique orientation or to a unique set of principal values. In molecular structural refinements against both X-ray and RDC data (possibly complemented by PCS data), there are several cases where constraining reciprocal orientation and/or magnitude of tensors may be useful. Examples that can be easily encountered can be grouped into two cases:
-
(a)
rigid systems composed of multiple subunits, where RDCs have been measured in different samples to reduce spectral complexity: in this case the reciprocal orientations of the tensors must be fixed, whereas their magnitude can vary because of slight differences in the concentration of the alignment medium;
-
(b)
rigid systems composed of multiple domains, where the relative orientations of the domains in solution are expected to be different from the solid state: in these cases the reciprocal orientations of the tensors must be allowed to vary, whereas their magnitude is fixed.
We have applied the new program for the refinement of three systems composed of multiple units belonging to these cases. In all three cases we have found that it is possible to fit the data to a single structural model, without invoking mobility to explain the inconsistency between the NMR data and the X-ray structures.
Methods
All refinement calculations were performed using REFMAC 5.9.0000 (Murshudov et al. 2011; Kovalevskiy et al. 2018), in which an option to constrain the relationship between different tensors have been introduced (see results and discussion for the details about implementation). The refinement protocol applied for the simultaneous refinement using NMR and X-ray is detailed in Rinaldelli et al. (2014).
In the most general form, the effect of residual dipolar coupling (RDC) is described as follows (Bertini et al. 2002b):
with
where rAB is the distance between the two coupled nuclei A and B, and sij are the components of the molecular alignment tensor. The anisotropies of the alignment tensor are described by the fraction of alignment along the z-axis (A) and by the rhombicity (η) as follows:
when \({\tilde {S}_{ii}}\) are the components of the alignment tensor in the frame in which it is diagonal.
In the case that the alignment of the molecule is induced by magnetic susceptibility anisotropy, the alignment is dictated by the anisotropy of the magnetic susceptibility tensor \(\varvec{\chi}\) and the tensor components are expressed as follows:
where \({\tilde {S}_{ii}}\) are the principal components of the alignment tensor S, \({\tilde {\chi }_{ii}}\) are the principal components of the magnetic susceptibility tensor \(\varvec{\chi}\), and \(\bar {\varvec{\chi}}\) is one-third of the trace of the tensor \(\varvec{\chi}\) (note that only the anisotropic components of the tensor should be considered), and it is common to define \(\Delta {\chi _{ax}}=\frac{3}{2}{\tilde {\chi }_{zz}}\) and \(\Delta {\chi _{rh}}=\frac{{{{\tilde {\chi }}_{xx}} - {{\tilde {\chi }}_{yy}}}}{2}\).
If the magnetic susceptibility anisotropy is induced by paramagnetic metals, PCSs are usually also measured. PCSs are related to the nuclear position according to the following equation (Bertini et al. 2002b):
Position and orientation of protein domains in solution and in the crystal state were compared as follows: the N-terminal domains of solution and crystal structures are superimposed and the distance between the C-terminal domains in the two states is evaluated in terms of the distance between their centres of mass, and of the angular deviation between their orientations. The latter is calculated as the angle θ obtained from the rotation matrix R which brings the C-terminal domain of one structure to be superimposed to the C-terminal domain of the other structure, according to the following equation:
The back-calculation of tensor parameters and the rigid body minimizations were performed using the FANTEN web application (Rinaldelli et al. 2015). Comparison of the tensors back-calculated independently for different structural units is done according to two criteria (three for paramagnetic cases): tensor magnitude, tensor alignment, and metal distance for paramagnetic cases (Russo et al. 2013). Comparison of the tensor magnitude is obtained from the ratio of their axial components of as following:
where values close to 1 indicate very similar tensor magnitude. Comparison of the tensor alignment (and shape) is performed by calculating the normalized dot product of the five independent components constituting the Saupe matrices (S) of the two tensors as following:
where values close to 1 indicate very similar tensor alignment. Metal distances are calculated as geometric distances between the average position of paramagnetic metals, as obtained after the refinement calculation, and the position of each lanthanoid as back-calculated from the C-terminal domain.
Experimental reflections are taken from the corresponding PDB entry, whereas the NMR data are available in the original publications.
Results and discussion
Program implementation
In the previous version of REFMAC-NMR (Rinaldelli et al. 2014), the alignment tensor S describing the RDCs (or the \(\varvec{\chi}\) tensor describing the PCSs) was determined through a Gauss–Newton optimization approach of 5 tensor components Szz, Sxx, Syy, Sxy, Sxz, Syz. Using these components as fitting parameters, application of constraints is rather involved. Therefore, we have re-parameterized the problem to allow for the inclusion of such constraints.
Instead of using the 5 tensor components given above, the tensor S is reconstructed as follows: the orientation is provided by three variables describing the rotation that brings the tensor S from any arbitrary molecular frame to the frame where it is diagonal, and the magnitude is determined by two of the diagonal elements, \({\tilde {S}_{xx}}\) and \({\tilde {S}_{yy}}\)\(\left( {{{\tilde {S}}_{zz}}= - \,{{\tilde {S}}_{xx}} - {{\tilde {S}}_{yy}},\,{\text{as the tensor is defined as traceless}}} \right)\). For a convenient sampling of the orientational space and to simplify the handling of the derivatives, the rotation is expressed in terms of quaternions:
where i, j, k are unit vectors representing the three Cartesian axes, \(v_1, v_2, v_3\) are the components of the unit vector defining the axis of rotation, and \(\theta\) is the angle of rotation. To represent a rotation, quaternions must satisfy the constraint \(\left| {\varvec{q}} \right|=1\) (unit quaternions). This reduces the number of independent variables describing the rotation matrix to three. Thus, the new parameters used in the calculations are the following:
The minimization performed by REFMAC for the structure refinement requires the computation of the first and second derivatives with respect to the selected parameters of the target function f to be minimized, reported in “Problem re-parameterization“ in Appendix.
In the presence of two structural units, two independent tensors can be defined, Sa and Sb, depending on the two sets of the parameters pa and pb defined as:
The function f is thus the sum of the functions referring to each domain:
where
The first and second derivatives of f are given by the derivatives of fa with respect to the pa parameters and of fb with respect to the pb parameters, and are reported in the “Imposing anisotropy and orientation constraints between tensors” in Appendix.
In order to constrain the orientation of the two tensors to be same (\({q_{a,i}}={q_{b,i}}\) for i = 0, 1, 2, 3), parameters and target function can be re-expressed in the following way:
where the non-null elements of the first and second derivatives of f for the present case are reported in “Imposing anisotropy and orientation constraints between tensors” in Appendix.
Likewise, in order to constrain the anisotropy values of the two tensors to be the same (\({\tilde {S}_{a,ii}}={\tilde {S}_{b,ii}}\) for ii = xx, yy), parameters and target function can be re-expressed in the following way:
where the non-null elements of the first and second derivatives of f are given in “Imposing anisotropy and orientation constraints between tensors” in Appendix.
Analogous considerations can be done for the tensor \(\varvec{\chi}\), used when PCSs are used.
The constraints described above have been implemented in REFMAC-NMR and can be applied to any generic set of tensors, according to the standard instruction file provided to REFMAC-NMR (see some examples in the Supplementary Materials). The algorithm can thus identify the tensors to be constrained (in orientation, anisotropy values, or both) according to user’s instructions, and perform the appropriate minimizations.
Using tensor constraints in REFMAC-NMR refinement
Tensor orientation in rigid multisubunit systems
To reduce the spectral complexity, the different subunits of a multisubunit systems may be separately expressed and reconstituted in vitro, labelling them differentially. This approach might introduce slight perturbations when recording RDCs with external alignment media: since the RDCs are collected in different samples, the concentration of the alignment medium may vary from one sample to the other, and this might simulate of protein mobility. We have recently encountered such a situation in the refinement of the complex between Sxl and CSD1 against X-ray and diamagnetic RDC data (Hennig et al. 2014; Carlon et al. 2016b). The refinement calculations were performed using a single alignment tensor for the complete RDC data and scaling the CSD1 RDC values by an empirical factor 0.8. This scaling factor was obtained from the comparison of the axial anisotropies obtained from the refinements performed for two proteins using two independent tensors.
Here we re-examine this system by imposing the tensors from the individual subunits to be equally oriented. A slightly different concentration of the alignment medium, in fact, affects the magnitude of the alignment tensors, but not their orientation. The inclusion of this orientation constraint represents the safest approach for an optimal refinement and permits the identification of the scaling factor between the different experimental conditions. We have thus refined again the structure of the complex between Sxl and CSD1 against the X-ray and diamagnetic RDC data using two alignment tensors constrained to have the same orientation.
The result of the refinements of the individual Sxl and CSD1 domains performed with either a single tensor (and uniform scaling of the CSD1 RDC values by an empirical factor 0.8), as previously done, or with the inclusion of the orientation constraint between the tensors of the two domains, now allowed by REFMAC-NMR, are compared in terms of REFMAC-NMR output and tensor comparisons (Table 1 and S1). A slight increase in the quality of the refinement, in terms of smaller R-free and Q-factor, is present when the orientation constraint is applied.
Tensor magnitude in rigid multidomain systems
There are cases in which a multidomain protein experiences different reciprocal arrangements of its domains upon binding to targets of different size. This is for instance the case of calmodulin (CaM) (Kursula 2014), the prototypical calcium sensor, upon binding to the death-associated protein kinase (DAPk, pdb code: 2X0G) (de Diego et al. 2010) or to a peptide that is derived from it (pdb code: 1YR5) (Bertini et al. 2009). DAPk is much bigger than CaM, whereas the fragment peptide is smaller. When crystallized in the peptide-bound form, the two domains of CaM experience crystal packing forces, which are mitigated when the crystallization occurs in the presence of the whole DAPk. For this system, paramagnetic NMR data (PCSs and RDCs) collected for CaM in the peptide-bound state were found in disagreement with the crystal structure. A solution structure was thus calculated, showing that the peptide-bound CaM adopts a more extended conformation in solution with respect to the crystal (Bertini et al. 2009). Differently, in the case of CaM bound to IQ peptide (pdb code: 2BE6), the agreement between the X-ray structure and paramagnetic NMR data is already quite remarkable (Russo et al. 2013). However, since three different conformations are observed in the crystals, in (Russo et al. 2013) the NMR data were analysed invoking interdomain mobility.
We here analyse the cases of CaM when bound to the DAPk peptide and to the IQ peptide again, through a join refinement including X-ray data and the available paramagnetic NMR data, and making use of the constraint that forces the tensors of the two domains to have the same magnitude and rhombicity, but allowing for different orientations. In this way, even if the domain arrangement in solution is different from the solid state, the PCS and RDC data can be reconciled with the X-ray data, because the different orientation of the tensors reports on the different relative orientation of the protein domains in solution. Following the REFMAC-NMR refinement, a rigid body minimization was performed [using FANTEN web application (Rinaldelli et al. 2015)] in order to recover the relative arrangement of the protein domains in solution (i.e., by matching orientation and origin of the tensors of the two domains). For the calculation of both the DAPk-peptide-bound CaM and DAPk-bond form the NMR data were the same, referring to the peptide-bound sample.
The quality of the refinements was evaluated in terms of (a) overall agreement with the experimental data (R-value, R-free, Q-factor PCS, Q-factor RDC) and with the ideal geometries (Tables 2, 3, 4) (Fig. 1); (b) agreement between experimental and back-calculated NMR data, in terms of residue-specific variations (Figs. S1–S3), and (c) agreement between the tensor parameters back-calculated for each domain (Fig. 2). The latter parameters, obtained with a best fit of the PCS and RDC data against each protein domain independently, reflect the tendency for the system to agree with a single rigid structure. For a single rigid structure, in fact, the magnitude and orientation of the tensors of the different domains should match each other and the positions of the paramagnetic metals should correspond. These quality factors were evaluated for the original X-ray structures (“X” in Figs. 1, 2), for the structures refined with a single tensor for each metal (“F” in Figs. 1, 2), and for the structures refined using the tensor magnitude constraint followed by rigid body minimization of domain positions (“SC” in Figs. 1, 2).

Agreement between observed and calculated data for the considered structures of CaM. Q-factors of PCSs and RDCs for the N- and C-terminal domains are calculated for the original X-ray structures (X), for the structures refined by REFMAC-NMR (F), and for the structures refined using the tensor magnitude constraint after applying rigid body minimization (SC)

Comparison between the tensor calculated independently for the N- and C-terminal domains in terms of tensor sizes, tensor alignments, and metal positions for the original X-ray structures (X), for the structures refined by REFMAC-NMR (F), and for the structures refined using the tensor magnitude constraint after applying rigid body minimization (SC)
The refinements for the three structures (1YR5, 2X0G and 2BE6) reveal some crucial different features.
Structure refinement of CaM in complex with DAPk
As previously found, the peptide-bound crystal structure 1YR5 is incompatible with the solution NMR data: even at the point where the agreement of X-ray and ideal geometry is barely acceptable (Table 2), a good fit of the NMR data cannot be obtained (Fig. 1, column “CaM-DAPk peptide”). Furthermore, the back-calculated tensor parameters indicate poor alignment between the tensors calculated for the C-terminal and for the N-terminal domain (Fig. 2, column “CaM-DAPk peptide”). The structure obtained after rigid body minimization becomes closer to the previously found solution structure 2K61 (see Fig. 3): the distance between the centres of mass of the C-terminal domains of the CaM-DAPk peptide structure and of the solution structure decreases from 6.4 to 4.6 Å after the use of rigid body minimization, and the angular deviation (as described in “Materials and methods”) is reduced from 17.1° to 11.5°.

Comparison between the original NMR structure of the peptide-bound CaM (2K61, blue) and a the X-ray structures 1YR5 (light red) and 2X0G (light green); b the refined 1YR5 (red) and 2X0G (green) structures after rigid body minimization
This overall disagreement is easily underpinned by the distribution of the residue-by-residue discrepancy between experimental and back-calculated data (Figs. S1a–S1c). As already reported in a previous work by some of us (Bertini et al. 2009), this is easily explained by the presence of large inter-protein contacts that are present in the crystal (Fig. 4, “CaM-DAPk peptide”). These contacts result to be mostly abolished in the protein-bound crystals (Fig. 4, “CaM-DAPk peptide”) and this is likely due to the larger size of the DAPk protein with respect to CaM and, consistently, the residue-by residue discrepancies are much smaller (Fig. S1a–S1c). As a result, the structure of CaM in complex with the whole DAPk protein results in much better agreement with the solution NMR data collected for the peptide-bound complex, as it is apparent from the quality of the joint refinement of 2X0G (Table 3) as well as from the agreement between the back-calculated tensors for the N- and C-terminal domains (Figs. 1 and 2, column “CaM-DAPk protein”). It is worth to note that the refined solution structure is very close to the previously found solution structure 2K61 (see Fig. 3): the distance between the centres of mass of the C-terminal domains of the CaM-DAPk protein structure and of the solution structure decreases from 5.7 to 2.3 Å after the use of rigid body minimization, and the angular deviation is reduced from 33.0° to 15.0°. As a final remark we note that a residual discrepancy is found between the back-calculated tensors of the N-terminal and C-terminal domains, mostly in the magnitude of the tensors and in the metal distance, especially for ytterbium(III). This residual discrepancy can be attributed to a limited but significant mobility as highlighted in (Andrałojć et al. 2014).

Visualization of intermolecular contacts in the crystal arrangements. PBD codes: a 1YR5, b 2X0G, c 2BE6. Protein residues are coloured according to the differences between experimental and back-calculated PCS values multiplied by r3 for the refined structures obtained after rigid body minimization
Structure refinement of CaM in complex with IQ peptide
In the case of CaM bound to IQ peptide, an extensive NMR dataset has been collected by the Griesinger’s group (Russo et al. 2013). The NMR data are in quite good agreement with each of the three monomers in the 2BE6 X-ray structure, but the back-calculated tensor parameters and the residue-specific differences between experimental and back-calculated data highlight some relevant discrepancies. These discrepancies were analysed in the original paper under the assumption that the complex in solution experiences some conformational variability. The experimental data were fit by an ensemble which encompasses the X-ray conformations together with a few selected MD-derived conformations.
We have found that a structural refinement using a single tensor for each metal, i.e.: assuming that the structure is not only rigid but also the same in the crystal and in solution (“F” in Fig. 2), is in relatively good agreement with the data (Table 4). However, some localized residue-specific discrepancies still remain, indicating that the fit is suboptimal. By applying the tensor magnitude constraint and thus allowing the two domains to be differently positioned in solution with respect to the solid state, the quality of the fit improves as well as the agreement between the back-calculated tensors (Fig. 2). The analysis of the residue-specific discrepancies also reveals that most of the biases have been removed (Figs. S3a–S3f). Again, the origin of the slightly different arrangement of the protein domains in solution is likely ascribable to the lack of the intermolecular interactions that connect each peptide-bound CaM with the neighbouring molecules in the crystal (Fig. 4). The small residual differences not accounted for by a single static model were similarly present even considering the NMR data as averaged over multiple X-ray conformations (Russo et al. 2013).
Conclusions
Only in a limited number of cases the crystal structures and the NMR data are smoothly in agreement with one another, but not all cases of inconsistency are necessarily significant. This is due to the intrinsic short-range inaccuracy of X-ray, especially in determining the position of hydrogen atoms, which are at the core of the NMR observations. Joint refinement of crystal structures against both X-ray experimental data and NMR data has thus proven a powerful tool to detect whether this inconsistency is real or if it is caused by the “structural noise”. To strengthen the use of the NMR data it is useful to impose constraints among the properties of the tensors (magnitude and/or orientation) to reduce the number of unknowns when different tensors need to be used, either because of structural rearrangement between the crystal and the solution or because different samples with differential labelling schemes are used to reduce the spectral complexity in the NMR measurements. We here describe the implementation of such a possibility in REFMAC-NMR, and show the efficiency of this approach in real-life cases. By these examples we demonstrate that using constraints among tensors is beneficial for improving the quality of the refinements even in cases where the experimental data are not sufficient to robustly estimate all the parameters. The changes in the structures are mostly involving heavy atoms and result in an overall improvement of the quality of the structure as evaluated by MolProbity (Chen et al. 2009, see Supplementary Material).
Change history
08 May 2019
The article “Joint X-ray/NMR structure refinement of multidomain/multisubunit systems” written by “Azzurra Carlon, Enrico Ravera, Giacomo Parigi, Garib N. Murshudov and Claudio Luchinat” was originally published electronically on the publisher’s internet portal (currently SpringerLink) on 11 October 2018 without open access.
References
Andrałojć W, Luchinat C, Parigi G, Ravera E (2014) Exploring regions of conformational space occupied by two-domain proteins. J Phys Chem B 118:10576–10587. https://doi.org/10.1021/jp504820w
Andrałojć W, Berlin K, Fushman D et al (2015) Information content of long-range NMR data for the characterization of conformational heterogeneity. J Biomol NMR 62:353–371. https://doi.org/10.1007/s10858-015-9951-6
Andrałojć W, Hiruma Y, Liu W-M et al (2017) Identification of productive and futile encounters in an electron transfer protein complex. Proc Natl Acad Sci USA 114:E1840–E1847. https://doi.org/10.1073/pnas.1616813114
Banci L, Bertini I, Bren KL et al (1996) The use of pseudocontact shifts to refine solution structures of paramagnetic metalloproteins: Met80Ala cyano-cytochrome c as an example. J Biol Inorg Chem 1:117–126
Banci L, Bertini I, Cremonini MA et al (1998a) PSEUDODYANA for NMR structure calculation of paramagnetic metalloproteins using torsion angle molecular dynamics. J Biomol NMR 12:553–557
Banci L, Bertini I, Huber JG et al (1998b) Partial orientation of oxidized and reduced cytochrome b5 at high magnetic fields: magnetic susceptibility anisotropy contributions and consequences for protein solution structure determination. J Am Chem Soc 120:12903–12909. https://doi.org/10.1021/ja981791w
Banci L, Bertini I, Cavallaro G et al (2004) Paramagnetism-based restraints for Xplor-NIH. J Biomol NMR 28:249–261
Berlin K, O’Leary DP, Fushman D (2010) Structural assembly of molecular complexes based on residual dipolar couplings. J Am Chem Soc 132:8961–8972. https://doi.org/10.1021/ja100447p
Berlin K, Castañeda CA, Schneidman-Duhovny D et al (2013) Recovering a representative conformational ensemble from underdetermined macromolecular structural data. J Am Chem Soc 135:16595–16609. https://doi.org/10.1021/ja4083717
Bertini I, Donaire A, Jiménez B et al (2001) Paramagnetism-based versus classical constraints: an analysis of the solution structure of Ca Ln calbindin D9k. J Biomol NMR 21:85–98
Bertini I, Luchinat C, Parigi G (2002a) Paramagnetic constraints: an aid for quick solution structure determination of paramagnetic metalloproteins. Concepts Magn Reson 14:259–286
Bertini I, Luchinat C, Parigi G (2002b) Magnetic susceptibility in paramagnetic NMR. Prog Nucl Magn Reson Spectrosc 40:249–273
Bertini I, Del Bianco C, Gelis I et al (2004) Experimentally exploring the conformational space sampled by domain reorientation in calmodulin. Proc Natl Acad Sci USA 101:6841–6846
Bertini I, Kursula P, Luchinat C et al (2009) Accurate solution structures of proteins from X-ray data and minimal set of NMR data: calmodulin peptide complexes as examples. J Am Chem Soc 131:5134–5144
Bertini I, Giachetti A, Luchinat C et al (2010) Conformational space of flexible biological macromolecules from average data. J Am Chem Soc 132:13553–13558
Brunger AT, Kuriyan J, Karplus M (1987) Crystallographic R factor refinement by molecular dynamics. Science 235:458–460
Carlon A, Ravera E, Andrałojć W et al (2016a) How to tackle protein structural data from solution and solid state: an integrated approach. Prog Nucl Magn Reson Spectrosc 92–93:54–70
Carlon A, Ravera E, Hennig J et al (2016b) Improved accuracy from Joint X-ray and NMR refinement of a protein–RNA complex structure. J Am Chem Soc 138:1601–1610. https://doi.org/10.1021/jacs.5b11598
Cerofolini L, Fields GB, Fragai M et al (2013) Examination of matrix metalloproteinase-1 in solution: a preference for the pre-collagenolysis state. J Biol Chem 288:30659–30671. https://doi.org/10.1074/jbc.M113.477240
Chao JA, Williamson JR (2004) Joint X-ray and NMR refinement of the yeast L30e-mRNA complex. Structure 12:1165–1176. https://doi.org/10.1016/j.str.2004.04.023
Chen VB, Arendall WB, Headd JJ et al (2009) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D 66:12–21. https://doi.org/10.1107/S0907444909042073
Chou JJ, Li S, Klee CB, Bax A (2001) Solution structure of Ca2+ calmodulin reveals flexible hand-like properties of its domains. Nat Struct Mol Biol 8:990–997
Clore GM (2011) Exploring sparsely populated states of macromolecules by diamagnetic and paramagnetic NMR relaxation. Protein Sci 20:229–246. https://doi.org/10.1002/pro.576
Cornilescu G, Marquardt J, Ottiger M, Bax A (1998) Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc 120:6836–6837
de Diego I, Kuper J, Bakalova N et al (2010) Molecular basis of the death-associated protein kinase-calcium/calmodulin regulator complex. Sci Signal 3:ra6–ra6. https://doi.org/10.1126/scisignal.2000552
Diaz-Moreno I, Diaz-Quintana A, De la Rosa MA, Ubbink M (2005) Structure of the complex between plastocyanin and cytochrome f from the cyanobacterium nostoc Sp. PCC 7119 as determined by paramagnetic NMR. J Biol Chem 280:18908–18915
Fenwick RB, van den Bedem H, Fraser JS, Wright PE (2014) Integrated description of protein dynamics from room-temperature X-ray crystallography and NMR. Proc Natl Acad Sci USA 111:E445–E454. https://doi.org/10.1073/pnas.1323440111
Fragai M, Luchinat C, Parigi G, Ravera E (2013) Conformational freedom of metalloproteins revealed by paramagnetism-assisted NMR. Coord Chem Rev 257:2652–2667. https://doi.org/10.1016/j.ccr.2013.02.009
Gaponenko V, Sarma SP, Altieri AS et al (2004) Improving the accuracy of NMR structures of large proteins using pseudocontact shifts as long/range restraints. J Biomol NMR 28:205–212
Gochin M, Roder H (1995) Protein structure refinement based on paramagnetic NMR shifts. applications to wild-type and mutants forms of cytochrome c. Protein Sci 4:296–305
Hass MA, Ubbink M (2014) Structure determination of protein–protein complexes with long-range anisotropic paramagnetic NMR restraints. Curr Opin Struct Biol 24:45–53. https://doi.org/10.1016/j.sbi.2013.11.010
Hennig J, Militti C, Popowicz GM et al (2014) Structural basis for the assembly of the Sxl–Unr translation regulatory complex. Nature 515:287–290. https://doi.org/10.1038/nature13693
Hoffman DW, Cameron CS, Davies C et al (1996) Ribosomal protein L9: a structure determination by the combined use of X-ray crystallography and NMR spectroscopy. J Mol Biol 264:1058–1071. https://doi.org/10.1006/jmbi.1996.0696
Jensen MR, Hansen DF, Ayna U et al (2006) On the use of pseudocontact shifts in the structure determination of metalloproteins. Magn Reson Chem 44:294–301
Knight MJ, Felli IC, Pierattelli R et al (2013) Magic angle spinning NMR of paramagnetic proteins. Acc Chem Res 46:2108–2116. https://doi.org/10.1021/ar300349y
Koehler J, Meiler J (2011) Expanding the utility of NMR restraints with paramagnetic compounds: background and practical aspects. Prog Nucl Magn Reson Spectrosc 59:360–389. https://doi.org/10.1016/j.pnmrs.2011.05.001
Kovalevskiy O, Nicholls RA, Long F, Carlon A, Murshudov GN (2018) Overview of refinement procedures within 5: utilizing data from different sources. Acta Crystallographica Section D Struct Biol 74(3):215–227
Kurland RJ, McGarvey BR (1970) Isotropic NMR shifts in transition metal complexes: calculation of the Fermi contact and pseudocontact terms. J Magn Reson 2:286–301
Kursula P (2014) The many structural faces of calmodulin: a multitasking molecular jackknife. Amino Acids 46:2295–2304. https://doi.org/10.1007/s00726-014-1795-y
Lange OF, Lakomek N-A, Farés C et al (2008) Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 320:1471–1475
Miller M, Lubkowski J, Mohana Rao JK et al (1996) The oligomerization domain of p53: crystal structure of the trigonal form. FEBS Lett 399:166–170. https://doi.org/10.1016/S0014-5793(96)01231-8
Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53:240–255. https://doi.org/10.1107/S0907444996012255
Murshudov GN, Skubák P, Lebedev AA et al (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D 67:355–367. https://doi.org/10.1107/S0907444911001314
Nitsche C, Otting G (2017) Pseudocontact shifts in biomolecular NMR using paramagnetic metal tags. Prog Nuclear Magn Reson Spectrosc 98–99:20–49. https://doi.org/10.1016/j.pnmrs.2016.11.001
Pintacuda G, John M, Su X-C, Otting G (2007) NMR structure determination of protein–ligand complexes by lanthanide labeling. Acc Chem Res 40:206–212. https://doi.org/10.1021/ar050087z
Ravera E, Parigi G, Luchinat C (2017) Perspectives on paramagnetic NMR from a life sciences infrastructure. J Magn Reson 282:154–169. https://doi.org/10.1016/j.jmr.2017.07.013
Raves ML, Doreleijer JF, Vis H et al (2001) Joint refinement as a tool for thorough comparison between NMR and X-ray data and structures of HU protein. J Biomol NMR 21:235–248
Rinaldelli M, Ravera E, Calderone V et al (2014) Simultaneous use of solution NMR and X-ray data in REFMAC5 for joint refinement/detection of structural differences. Acta Crystallogr D 70:958–967. https://doi.org/10.1107/S1399004713034160
Rinaldelli M, Carlon A, Ravera E et al (2015) FANTEN: a new web-based interface for the analysis of magnetic anisotropy-induced NMR data. J Biomol NMR 61:21–34. https://doi.org/10.1007/s10858-014-9877-4
Russo L, Maestre-Martinez M, Wolff S et al (2013) Interdomain dynamics explored by paramagnetic NMR. J Am Chem Soc 135:17111–17120. https://doi.org/10.1021/ja408143f
Schiffer CA, Huber R, Wüthrich K, van Gunsteren WF (1994) Simultaneous refinement of the structure of BPTI against NMR data measured in solution and X-ray diffraction data measured in single crystals. J Mol Biol 241:588–599. https://doi.org/10.1006/jmbi.1994.1533
Schlundt A, Tants J-N, Sattler M (2017) Integrated structural biology to unravel molecular mechanisms of protein-RNA recognition. Methods 118–119:119–136. https://doi.org/10.1016/j.ymeth.2017.03.015
Schmitz C, Bonvin AMJJ (2011) Protein-protein HADDocking using exclusively pseudocontact shifts. J Biomol NMR 50:263–266. https://doi.org/10.1007/s10858-011-9514-4
Schmitz C, Vernon R, Otting G et al (2012) Protein structure determination from pseudocontact shifts using ROSETTA. J Mol Biol 416:668–677. https://doi.org/10.1016/j.jmb.2011.12.056
Schwieters CD, Kuszewski JJ, Tjandra N, Marius Clore G (2003) The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160:65–73. https://doi.org/10.1016/S1090-7807(02)00014-9
Shaanan B, Gronenborn A, Cohen G et al (1992) Combining experimental information from crystal and solution studies: joint X-ray and NMR refinement. Science 257:961–964. https://doi.org/10.1126/science.1502561
Simon B, Madl T, Mackereth CD et al (2010) An efficient protocol for NMR-spectroscopy-based structure determination of protein complexes in solution. Angew Chem Int Ed Engl 49:1967–1970. https://doi.org/10.1002/anie.200906147
Tang C, Schwieters CD, Clore GM (2007) Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature 449:1078–1082. https://doi.org/10.1038/nature06232
Tang M, Sperling LJ, Berthold DA et al (2011) High-resolution membrane protein structure by joint calculations with solid-state NMR and X-ray experimental data. J Biomol NMR 51:227–233. https://doi.org/10.1007/s10858-011-9565-6
Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH (1995) Nuclear magnetic dipole interactions in field-oriented proteins: information for structure determination in solution. Proc Natl Acad Sci USA 92:9279–9283
Valafar H, Prestegard JH (2004) REDCAT: a residual dipolar coupling analysis tool. J Magn Reson 167:228–241. https://doi.org/10.1016/j.jmr.2003.12.012
van den Bedem H, Fraser JS (2015) Integrative, dynamic structural biology at atomic resolution–it’s about time. Nat Methods 12:307–318. https://doi.org/10.1038/nmeth.3324
Volkov AN, Worrall JAR, Holtzmann E, Ubbink M (2006) Solution structure and dynamics of the complex between cytochrome c and cytochrome c peroxidase determined by paramagnetic NMR. Proc Natl Acad Sci USA 103:18945–18950. https://doi.org/10.1073/pnas.0603551103
Ward AB, Sali A, Wilson IA (2013) Integrative structural biology. Science 339:913–915. https://doi.org/10.1126/science.1228565
Winn MD, Ballard CC, Cowtan KD et al (2011) Overview of the CCP 4 suite and current developments. Acta Crystallogr Sect D 67:235–242. https://doi.org/10.1107/S0907444910045749
Zweckstetter M, Bax A (2002) Evaluation of uncertainty in alignment tensors obtained from dipolar couplings. J Biomol NMR 23:127–137
Acknowledgements
The support from Fondazione Cassa di Risparmio di Firenze, MIUR PRIN 2012SK7ASN, the European Commission projects iNEXT No. 653706, West-Life No. 675858, EMBO ASTF 620–2015, and Instruct-ERIC, a Landmark ESFRI project, and specifically the CERM/CIRMMP Italy center, is acknowledged. GNM is funded by Medical Research Council (Grant No. MC_UP_A025_1012). Extensive and frank discussions with Christian Griesinger on how much information on dynamics can be reliably extracted from the NMR data of CaM-IQ are acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original version of this article was revised due to a retrospective Open Access order.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix
Appendix
Problem re-parameterization
To allow for the inclusion of the tensor orientation and magnitude constraints, it is convenient to re-express the problem in terms of eigenvectors and eigenvalues of the alignment tensor S. Eigenvectors ex, ey, ez and associated eigenvalues \({\tilde {S}_{xx}}\), \({\tilde {S}_{yy}}\), \({\tilde {S}_{zz}}\) encode, respectively, the information about the orientation and the magnitude/anisotropy of the tensor S, and are related to the tensor matrix by the simple relationship:
Eigenvectors basis constitutes the rotation matrix R that transforms the tensor from the molecular frame to its principal axis frame. This allows expressing each element \({S_{ij}}\) of the alignment tensor S in terms of its principal components \({\tilde {S}_{ii}}\) and of element of the rotation matrix R as following:
Each element of the rotation matrix R is then more conveniently expressed in terms of quaternions, thus reducing the number of parameters describing the rotation from nine to four.
Given f the target function to be minimized, x the set of parameters for the original problem, defined as:
and p the set of parameters for the re-parameterized problem, defined in this particular case as:
the first and second derivatives of the re-parameterized problem can then be directly calculated starting from the derivatives of the original problem by application of the chain rule, as following:
where the last term of the second derivative has not be considered since it implies the use of the first derivative with the respect of the original problem, making the optimization less stable.
Imposing anisotropy and orientation constraints between tensors
In the presence of two structural units, two independent tensors can be defined, Sa and Sb, depending on the two sets of the parameters pa and pb defined as:
The function f is thus the sum of the functions referring to each domain:
where
The first and second derivatives of f are given by the derivatives of fa with respect to the pa parameters and of fb with respect to the pb parameters. The derivatives are reported, for simplicity, in the following matrix form where only the non-null elements are shown:
In order to constrain the orientation of the two tensors to be the same (\({q_{a,i}}={q_{b,i}}\,for\,i\,=\,0,{\text{ }}1,{\text{ }}2,{\text{ }}3\)), parameters and target function are expressed in the following way:
where the non-null elements of the first and second derivatives of f are given as following:
In order to constrain the anisotropy values of the two tensors to be the same (\({\tilde {S}_{a,ii}}={\tilde {S}_{b,ii}}\) for ii = xx, yy), parameters and target function can be re-expressed in the following way:
where the non-null elements of the first and second derivatives of f are given as following:
This approach can be used to perform the minimization of a generic number of tensors, constrained to each other by their anisotropy, orientation or both.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Carlon, A., Ravera, E., Parigi, G. et al. Joint X-ray/NMR structure refinement of multidomain/multisubunit systems. J Biomol NMR 73, 265–278 (2019). https://doi.org/10.1007/s10858-018-0212-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-018-0212-3