
Abstract
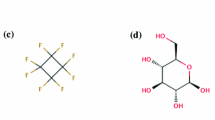
Hydration free energy calculations in explicit solvent have become an integral part of binding free energy calculations and a valuable test of force fields. Most of these simulations follow the conventional norm of keeping edge length of the periodic solvent box larger than twice the Lennard-Jones (LJ) cutoff distance, with the rationale that this should be sufficient to keep the interactions between copies of the solute to a minimum. However, for charged solutes, hydration free energies can exhibit substantial box size-dependence even at typical box sizes. Here, we examine whether similar size-dependence affects hydration of neutral molecules. Thus, we focused on two strongly polar molecules with large dipole moments, where any size-dependence should be most pronounced, and determined how their hydration free energies vary as a function of simulation box size. In addition to testing a variety of simulation box sizes, we also tested two LJ cut-off distances, 0.65 and 1.0 nm. We show from these simulations that the calculated hydration free energy is independent of the box-size as well as the LJ cut-off distance, suggesting that typical hydration free energy calculations of neutral compounds indeed need not be particularly concerned with finite-size effects as long as standard good practices are followed.




Similar content being viewed by others

References
Bennett CH (1976) Efficient estimation of free energy differences from Monte Carlo data. J Comput Phys 22(2):245–268
Beutler TC, Mark AE, van Schaik RC, Gerber PR, van Gunsteren WF (1994) Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem Phys Lett 222(6):529–539
Hünenberger PH, Reif MM (2011) Single-ion solvation: experimental and theoretical approaches to elusive thermodynamic quantities, vol 3, 1st edn. Royal Society of Chemistry, London
Jakalian A, Bush BL, Jack DB, Bayly CI (2000) Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem 21(2):132–146
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79(2):926–935
Kastenholz MA, Hünenberger PH (2006) Computation of methodology-independent ionic solvation free energies from molecular simulations. II. The hydration free energy of the sodium cation. J Chem Phys 124(22):224501
Mobley DL, Dumont É, Chodera JD, Dill KA (2007) Comparison of charge models for fixed-charge force fields: small-molecule hydration free energies in explicit solvent. J Phys Chem B 111(9):2242–2254
Mobley DL, Liu S, Cerutti DS, Swope WC, Rice JE (2011) Alchemical prediction of hydration free energies for SAMPL. J Comput Aided Mol Des 26(5):551–562
Reif MM, Oostenbrink C (2013) Net charge changes in the calculation of relative ligand-binding free energies via classical atomistic molecular dynamics simulation. J Comput Chem 35(3):227–243
Rocklin GJ, Mobley DL, Dill KA, Hünenberger PH (2013) Calculating the binding free energies of charged species based on explicit-solvent simulations employing lattice-sum methods: An accurate correction scheme for electrostatic finite-size effects. J Chem Phys 139(18):184103
Shirts M, Mobley D (2013) An introduction to best practices in free energy calculations. In: Monticelli L, Salonen E (eds.) Biomolecular Simulations, Methods in Molecular Biology, vol. 924, pp. 271–311. Humana Press
Shirts MR, Chodera JD (2008) Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys 129(12):124,105
Shirts MR, Mobley DL, Chodera JD (2007) Alchemical Free Energy Calculations: Ready for Prime Time?, vol. 3, chap. 4, pp. 41–59. Elsevier
Shirts MR, Mobley DL, Chodera JD, Pande VS (2007) Accurate and efficient corrections for missing dispersion interactions in molecular simulations. J Phys Chem B 111(45):13,052–13,063
Wang J, Wang W, Kollman PA, Case DA (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 25(2):247–260
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general AMBER force field. J Comput Chem 25(9):1157–1174
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Parameswaran, S., Mobley, D.L. Box size effects are negligible for solvation free energies of neutral solutes. J Comput Aided Mol Des 28, 825–829 (2014). https://doi.org/10.1007/s10822-014-9766-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-014-9766-7