
Abstract
In conjunction with the recent American Chemical Society symposium titled “Docking and Scoring: A Review of Docking Programs” the performance of the DOCK6 program was evaluated through (1) pose reproduction and (2) database enrichment calculations on a common set of organizer-specified systems and datasets (ASTEX, DUD, WOMBAT). Representative baseline grid score results averaged over five docking runs yield a relatively high pose identification success rate of 72.5 % (symmetry corrected rmsd) and sampling rate of 91.9 % for the multi site ASTEX set (N = 147) using organizer-supplied structures. Numerous additional docking experiments showed that ligand starting conditions, symmetry, multiple binding sites, clustering, and receptor preparation protocols all affect success. Encouragingly, in some cases, use of more sophisticated scoring and sampling methods yielded results which were comparable (Amber score ligand movable protocol) or exceeded (LMOD score) analogous baseline grid-score results. The analysis highlights the potential benefit and challenges associated with including receptor flexibility and indicates that different scoring functions have system dependent strengths and weaknesses. Enrichment studies with the DUD database prepared using the SB2010 preparation protocol and native ligand pairings yielded individual area under the curve (AUC) values derived from receiver operating characteristic curve analysis ranging from 0.29 (bad enrichment) to 0.96 (good enrichment) with an average value of 0.60 (27/38 have AUC ≥ 0.5). Strong early enrichment was also observed in the critically important 1.0–2.0 % region. Somewhat surprisingly, an alternative receptor preparation protocol yielded comparable results. As expected, semi-random pairings yielded poorer enrichments, in particular, for unrelated receptors. Overall, the breadth and number of experiments performed provide a useful snapshot of current capabilities of DOCK6 as well as starting points to guide future development efforts to further improve sampling and scoring.












Similar content being viewed by others
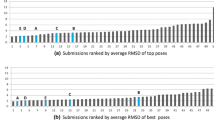
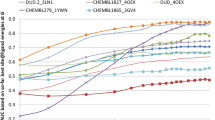
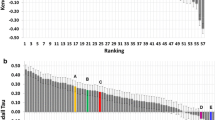
References
Klebe G (2006) Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today 11(13–14):580–594
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3(11):935–949
Kuntz ID (1992) Structure-based strategies for drug design and discovery. Science 257(5073):1078–1082
Jorgensen WL (2004) The many roles of computation in drug discovery. Science 303(5665):1813–1818
Shoichet BK (2004) Virtual screening of chemical libraries. Nature 432(7019):862–865
Irwin JJ, Shoichet BK (2004) ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model 45(1):177–182
Cross JB, Thompson DC, Rai BK, Baber JC, Fan KY, Hu Y, Humblet C (2009) Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J Chem Inf Model 49(6):1455–1474
Mukherjee S, Balius TE, Rizzo RC (2010) Docking validation resources: protein family and ligand flexibility experiments. J Chem Inf Model 50(11):1986–2000
Huang N, Shoichet BK, Irwin JJ (2006) Benchmarking sets for molecular docking. J Med Chem 49(23):6789–6801
Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE (1982) A geometric approach to macromolecule-ligand interactions. J Mol Biol 161(2):269–288
DesJarlais RL, Seibel GL, Kuntz ID, Furth PS, Alvarez JC, Ortiz de Montellano PR, DeCamp DL, Babé LM, Craik CS (1990) Structure-based design of nonpeptide inhibitors specific for the human immunodeficiency virus 1 protease. Proc Nat Acad Sci 87(17):6644–6648
Ewing TJA, Kuntz ID (1997) Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem 18(9):1175–1189
Ewing TJA, Makino S, Skillman AG, Kuntz ID (2001) DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des 15(5):411–428
Meng EC, Shoichet BK, Kuntz ID (1992) Automated docking with grid-based energy evaluation. J Comput Chem 13(4):505–524
Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC (2006) Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des 20(10):601–619
Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID (2009) DOCK 6: combining techniques to model RNA-small molecule complexes. RNA 15(6):1219–1230
Zou X, Yaxiong S, Kuntz ID (1999) Inclusion of solvation in ligand binding free energy calculations using the generalized-born model. J Am Chem Soc 121(35):8033–8043
Liu H-Y, Kuntz ID, Zou X (2004) Pairwise GB/SA scoring function for structure-based drug design. J Phys Chem B 108(17):5453–5462
Grant JA, Pickup BT, Nicholls A (2001) A smooth permittivity function for Poisson–Boltzmann solvation methods. J Comput Chem 22(6):608–640
Graves AP, Shivakumar DM, Boyce SE, Jacobson MP, Case DA, Shoichet BK (2008) Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J Mol Biol 377(3):914–934
Balius TE, Mukherjee S, Rizzo RC (2011) Implementation and evaluation of a docking-rescoring method using molecular footprint comparisons. J Comput Chem 32(10):2273–2289
SBU DOCK Tutorials. http://ringo.ams.sunysb.edu/index.php/DOCK_Tutorials. Last accessed Mar 01, 2012
UCSF DOCK Tutorials. http://dock.compbio.ucsf.edu/DOCK_6/tutorials/index.htm. Last accessed Mar 01, 2012
Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WTM, Mortenson PN, Murray CW (2007) Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem 50(4):726–741
Good A, Oprea T (2008) Optimization of CAMD techniques 3. Virtual screening enrichment studies: a help or hindrance in tool selection? J Comput Aided Mol Des 22(3):169–178
Kuhn HW (1955) The Hungarian method for the assignment problem. Nav Res Logist Quart 2:83–97
Munkres J (1957) Algorithms for the assignment and transportation problems. J Soc Ind Appl Math 5(1):32–38
Raymond JW, Willett P (2002) Maximum common subgraph isomorphism algorithms for the matching of chemical structures. J Comput Aided Mol Des 16(7):521–533
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612
Triballeau N, Acher F, Brabet I, Pin JP, Bertrand HO (2005) Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem 48(7):2534–2547
Jain A, Nicholls A (2008) Recommendations for evaluation of computational methods. J Comput Aided Mol Des 22(3):133–139
Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA (1998) Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate—DNA helices. J Am Chem Soc 120(37):9401–9409
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33(12):889–897
Rastelli G, Rio AD, Degliesposti G, Sgobba M (2010) Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem 31(4):797–810
Kuhn B, Gerber P, Schulz-Gasch T, Stahl M (2005) Validation and use of the MM-PBSA approach for drug discovery. J Med Chem 48(12):4040–4048
Thompson DC, Humblet C, Joseph-McCarthy D (2008) Investigation of MM-PBSA rescoring of docking poses. J Chem Inf Model 48(5):1081–1091
Hou T, Wang J, Li Y, Wang W (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 51(1):69–82
Macke TJ, Case DA (1997) Modeling unusual nucleic acid structures. ACS Sym Ser 682:379–393
Kolossvary I, Guida WC (1996) Low mode search. An efficient, automated computational method for conformational analysis: application to cyclic and acyclic alkanes and cyclic peptides. J Am Chem Soc 118(21):5011–5019
Kolossvary I, Keseru GM (2001) Hessian-free low-mode conformational search for large-scale protein loop optimization: application to c-jun N-terminal kinase JNK3. J Comput Chem 22(1):21–30
Sheridan RP, McGaughey GB, Cornell WD (2008) Multiple protein structures and multiple ligands: effects on the apparent goodness of virtual screening results. J Comput Aided Mol Des 22(3–4):257–265
Truchon JF, Bayly CI (2007) Evaluating virtual screening methods: good and bad metrics for the “early recognition” problem. J Chem Inf Model 47(2):488–508
Onufriev A, Bashford D, Case DA (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55(2):383–394
Weiser J, Shenkin PS, Still WC (1999) Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comput Chem 20(2):217–230
Hawkins GD, Cramer CJ, Truhlar DG (1996) Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J Phys Chem 100(51):19824–19839
Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65(3):712–725
Gasteiger J, Marsili M (1980) Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 36(22):3219–3228
Maignan S, Guilloteau JP, Pouzieux S, Choi-Sledeski YM, Becker MR, Klein SI, Ewing WR, Pauls HW, Spada AP, Mikol V (2000) Crystal structures of human factor Xa complexed with potent inhibitors. J Med Chem 43(17):3226–3232
Nar H, Bauer M, Schmid A, Stassen JM, Wienen W, Priepke HW, Kauffmann IK, Ries UJ, Hauel NH (2001) Structural basis for inhibition promiscuity of dual specific thrombin and factor Xa blood coagulation inhibitors. Structure 9(1):29–37
Chachra R, Rizzo RC (2008) Origins of resistance conferred by the R292K neuraminidase mutation via molecular dynamics and free energy calculations. J Chem Theory Comput 4(9):1526–1540
Acknowledgments
Greg Warren, Neysa Nevins, and Georgia McGauhey are thanked for organizing the special Docking and Scoring symposium. William J. Allen and Jiangyang Liu are thanked for code development and Steve Skiena is thanked for helpful discussions regarding implementation of symmetry corrected rmsd using the Hungarian matching algorithm. This work was supported in part by NIH grants GM57513 (D.A.C.), R01GM083669 (R.C.R.), and F31CA134201 (T.E.B.), as well as the Stony Brook University Office of the Vice President for Research and the New York State Office of Science Technology and Academic Research (NYSTAR). S.R.B. gratefully acknowledges the use of computational facilities at the Ohio Supercomputer Center and thanks OpenEye Scientific Software for an academic license. This work also used resources at the New York Center for Computational Sciences at Stony Brook University/Brookhaven National Laboratory supported by the US Department of Energy under Contract No. DE-AC02-98CH10886 and by the State of New York. Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institutes of Health (National Center for Research Resources grant 2P41RR001081, National Institute of General Medical Sciences grant 9P41GM103311).
Author information
Authors and Affiliations
Corresponding author
Additional information
Scott R. Brozell and Sudipto Mukherjee contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Brozell, S.R., Mukherjee, S., Balius, T.E. et al. Evaluation of DOCK 6 as a pose generation and database enrichment tool. J Comput Aided Mol Des 26, 749–773 (2012). https://doi.org/10.1007/s10822-012-9565-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-012-9565-y