
Abstract
Purpose
Bardet–Biedl syndrome (BBS: OMIM 209,900) is a rare ciliopathic human genetic disorder that affects many parts of the body systems. BBS is a genetically heterogeneous disorder with a wide spectrum of clinical manifestations which makes its diagnosis and management more challenging. RetNet reports 18 genes that cause BBS and each of genes has had several known mutations. Genetic studies suggesting that serologically defined colon cancer antigen 8 (SDCCAG8) gene mutations are a major cause of BBS.
Materials and methods
In this section, we investigated the consanguineous Iranian family members with BBS. Whole-exome sequencing and Sanger sequencing, were performed to screen and confirm the suspicious pathogenic mutations. The identified mutation was investigated using bioinformatics tools to predict the effect of the mutation on protein structure.
Results
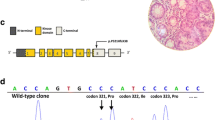
Sequential analysis identified a novel splice site mutation c.1221 + 2 T > A in the SDCCAG8 gene in BBS patients. Structure-based approaches have predicted significant structural alterations in SDCCAG8 protein.
Conclusions
This study was conducted to show the aberrant alternative splicing as one of the single splicing mutations identified can cause BBS by affecting the function of SDCCAG8 protein.





Similar content being viewed by others


Abbreviations
- BBS:
-
Bardet–Biedl syndrome
- SDCCAG8:
-
Serologically defined colon cancer antigen 8
- WES:
-
Whole-exome sequencing
References
Abdulla AB, Niloy AA, Shah TA, Biswas SK, Imran AK, Murshed KM et al (2009) Laurence Moon Bardet Biedl syndrome. Mymensingh Med J MMJ 18(1 Suppl):S124–S128
Badano JL, Leitch CC, Ansley SJ, May-Simera H, Lawson S, Lewis RA et al (2006) Dissection of epistasis in oligogenic Bardet–Biedl syndrome. Nature 439(7074):326–330
Tobin JL, Beales PL (2009) The nonmotile ciliopathies. Genet Med 11(6):386–402
Cherian MP, Al-Sanna'a NA (2009) Clinical spectrum of Bardet-Biedl syndrome among four Saudi Arabian families. Clin Dysmorphol 18(4):188–194
Schachat AP, Maumenee IH (1982) Bardet–Biedl syndrome and related disorders. Arch Ophthalmol (Chicago, IL: 1960) 100(2):285–8
Abu-Safieh L, Al-Anazi S, Al-Abdi L, Hashem M, Alkuraya H, Alamr M et al (2012) In search of triallelism in Bardet–Biedl syndrome. Eur J Hum Genet EJHG 20(4):420–427
Ajmal M, Khan MI, Neveling K, Tayyab A, Jaffar S, Sadeque A et al (2013) Exome sequencing identifies a novel and a recurrent BBS1 mutation in Pakistani families with Bardet–Biedl syndrome. Mol Vis 19:644–653
Chiang AP, Beck JS, Yen HJ, Tayeh MK, Scheetz TE, Swiderski RE et al (2006) Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc Natl Acad Sci USA 103(16):6287–6292
Abu Safieh L, Aldahmesh MA, Shamseldin H, Hashem M, Shaheen R, Alkuraya H et al (2010) Clinical and molecular characterisation of Bardet–Biedl syndrome in consanguineous populations: the power of homozygosity mapping. J Med Genet 47(4):236–241
Yates B, Braschi B, Gray KA, Seal RL, Tweedie S, Bruford EA (2017) Genenames.org: the HGNC and VGNC resources in 2017. Nucleic acids Res 45(D1):D619–D625
Hildebrandt F, Zhou W (2007) Nephronophthisis-associated ciliopathies. J Am Soc Nephrol JASN 18(6):1855–1871
Schaefer E, Zaloszyc A, Lauer J, Durand M, Stutzmann F, Perdomo-Trujillo Y et al (2011) Mutations in SDCCAG8/NPHP10 cause Bardet–Biedl syndrome and are associated with penetrant renal disease and absent polydactyly. Mol Syndromol 1(6):273–281
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS et al (2003) Human gene mutation database (HGMD): 2003 update. Hum Mutat 21(6):577–581
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics (Oxford, England) 25(9):1189–1191
Chojnacki S, Cowley A, Lee J, Foix A, Lopez R (2017) Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res 45(W1):W550–W553
Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics (Oxford, England) 27(3):343–350
Bertoni M, Kiefer F, Biasini M, Bordoli L, Schwede T (2017) Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep 7(1):10480
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5(4):725–738
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12(1):7–8
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinform 9:40
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R et al (2019) VarSome: the human genomic variant search engine. Bioinformatics (Oxford, England) 35(11):1978–1980
Fulton AB, Hansen RM, Glynn RJ (1993) Natural course of visual functions in the Bardet-Biedl syndrome. Arch Ophthalmol (Chicago, IL: 1960) 111(11):1500–1506
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA (1999) New criteria for improved diagnosis of Bardet–Biedl syndrome: results of a population survey. J Med Genet 36(6):437–446
Katsanis N, Lupski JR, Beales PL (2001) Exploring the molecular basis of Bardet-Biedl syndrome. Hum Mol Genet 10(20):2293–2299
Priya S, Nampoothiri S, Sen P, Sripriya S (2016) Bardet-Biedl syndrome: Genetics, molecular pathophysiology, and disease management. Indian J Ophthalmol 64(9):620–627
O'Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J (1996) The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis 27(6):776–783
Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, Stoetzel C et al (2010) Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet 42(10):840–850
Hildebrandt F, Attanasio M, Otto E (2009) Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol JASN 20(1):23–35
Airik R, Slaats GG, Guo Z, Weiss AC, Khan N, Ghosh A et al (2014) Renal-retinal ciliopathy gene Sdccag8 regulates DNA damage response signaling. J Am Soc Nephrol JASN 25(11):2573–2583
Anna A, Monika G (2018) Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet 59(3):253–268
de Calais FL, Smith LD, Raponi M, Maciel-Guerra AT, Guerra-Junior G, de Mello MP et al (2017) A study of splicing mutations in disorders of sex development. Sci Rep 7(1):16202
Acknowledgements
This study has been granted by the University of Medical Sciences, Tabriz, Iran (Grant Number: 64764). We would like to thank the University of Medical Sciences Semnan, Iran for their collaboration and support. Moreover, we would like to express our sincere thanks to the family members who participated in this study, and we profoundly appreciate their collaboration in making this study possible.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bahmanpour, Z., Daneshmandpour, Y., Kazeminasab, S. et al. A novel splice site mutation in the SDCCAG8 gene in an Iranian family with Bardet–Biedl syndrome. Int Ophthalmol 41, 389–397 (2021). https://doi.org/10.1007/s10792-020-01588-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10792-020-01588-x