
Abstract
Acute lung injury (ALI) caused by septic stimuli is still a major problem in critical care patients. We have shown previously that hydrogen sulfide (H2S) mediates anti-inflammatory and lung protective effects. In the present study, we aimed to investigate the underlying mechanisms. C57BL/6N mice were instilled with lipopolysaccharide (LPS) intranasally in the absence or presence of inhaled H2S for 6 h. LPS instillation led to alveolar wall thickening, an elevated ALI score, increased neutrophil transmigration, and elevated interleukin-1β cytokine release into the bronchoalveolar lavage fluid. In contrast, H2S inhalation prevented lung injury and inflammation despite LPS treatment. Moreover, H2S inhalation significantly inhibited protein expression of cystathionine-β-synthetase, heat shock protein 70, phosphorylated p38 MAP kinase, NADPH oxidase 2, and the formation of reactive oxygen species (ROS) in LPS-challenged animals. In conclusion, H2S prevents LPS-induced ALI by inhibition of pro-inflammatory and oxidative responses via the concerted attenuation of stress protein, MAP kinase, and ROS signaling pathways.
Similar content being viewed by others
BACKGROUND
Pulmonary infection is the most important cause of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) and continues to produce a high rate of morbidity and mortality in the intensive care unit [1]. Lung injury upon infection is characterized by a strong inflammatory response, reflected by transmigration of immune-competent cells (e.g., neutrophils) and the release of pro-inflammatory cytokines (e.g., interleukin-1β). Subsequently, the inflammatory process damages alveolar architecture resulting in critical impairment of lung function [2]. Despite antibiotic treatment and protective mechanical ventilation, to date no specific treatment has been shown to improve the clinical outcome of patients suffering from ALI/ARDS.
Several signaling pathways are involved in the outcome of ALI caused by infective stimuli. Among them, the induction of heat shock proteins like hemeoxygenase-1 (HO-1) and heat shock protein 70 (HSP70) [3], or the inhibition of MAP kinase signaling, all mediate cytoprotective and anti-inflammatory effects in various models of lung injury [4,5,6]. In addition, the inflammatory response in sepsis is also triggered by oxidative stress resulting from the excessive production of reactive oxygen species (ROS). In this respect, hydrogen sulfide (H2S) is an endogenously produced gasotransmitter that is known for its anti-inflammatory and redox regulating capacity. Exogenous administration of H2S has been shown to mediate protective effects in various animal disease models, such as ischemia-reperfusion injury [7], ventilator-induced ALI [8], hyperoxia-induced ALI [9], or oleic acid-induced lung injury [10]. We have recently demonstrated that inhalation of 80 parts per million (ppm) H2S exerts anti-inflammatory effects and can protect against ALI in an inflammatory model of lipopolysaccharide (LPS)-induced lung injury [11]. However, the molecular signaling pathways involved in the observed protection are widely unknown. We therefore hypothesized that H2S may modulate the generation of ROS and that H2S producing enzymes, heat shock protein responses, MAP kinase signaling, and the release and attraction of inflammatory mediators may also be affected by H2S in LPS-induced ALI. In the present study, we thus defined the role of anti-inflammatory, anti-oxidative, and feedback pathways in H2S-mediated lung protection during inflammation. We show for the first time that prevention of lung injury is associated with the anti-oxidative effects of H2S inhalation leading to a concerted inhibition of p38 MAPK signaling, ROS formation, and Nox2 expression.
METHODS
Animals and Experimental Setting
C57BL/6N mice (n = 24) were randomly assigned to four experimental groups: Group 1 (control): mice were exposed to synthetic air for 1 h, were instilled with 70 μl endotoxin-free PBS intranasally (i.n.), and breathed air for another 6 h. Group 2 (control + H2S): mice breathed air supplemented with 80 ppm H2S for 1 h, received PBS i.n., and breathed H2S for another 6 h. Group 3 (LPS): mice breathed air for 1 h, received 0.25 ng LPS in 70 μl PBS i.n. (Escherichia coli 055:B5; Sigma-Aldrich Chemie GmbH, Munich, Germany), and breathed air for another 6 h. Group 4 (LPS + H2S): mice breathed 80 ppm H2S for 1 h, received LPS i.n., and breathed H2S for another 6 h. Experiments were performed in a sealed Plexiglass chamber in which H2S concentration was measured continuously using a portable gas monitor (MX6 iBrid, Industrial Scientific Corporation, Oakdale, PA). All animal experiments were performed in accordance with guidelines of the local animal care commission (Ethics Committee University of Freiburg and Regierungspräsidium Freiburg, Freiburg, Germany, Permission No. G-07/25) and in conformance with the journals’ requirements for human and animal trials.
End of Experiment, Tissue Sampling, and Bronchoalveolar Lavage
At the end of each experiment, all mice were sacrificed by an intraperitoneal, overdosed injection of ketamine (180 mg/kg) and acepromazine (1.8 mg/kg). Bronchoalveolar lavage (BAL) fluid and lung tissue for Western Blot analysis and histological examination were gained as described previously [9, 11].
Cytokine Measurements
BAL aliquots were analyzed using an interleukin-1β (IL-1β) ELISA kit (R&D Systems GmbH, Wiesbaden, Germany) according to the manufacturers’ instructions.
Histological Examination
Cryosections of the left lung lobes and hematoxylin and eosin staining was performed and analyzed as described previously [9].
Detection of Reactive Oxygen Species
Dihydroethidium (DHE, Life Technologies GmbH, Darmstadt, Germany) was used to detect reactive oxygen species (ROS) in lung tissue as described earlier [9]. Densitometric analysis was performed with the ImageJ software (National Institutes of Health; Bethesda, MD; available at: http://rsbweb.nih.gov.).
Immunoblotting
Western blotting was performed as described recently [12]. The membranes were incubated with antibodies against 3-mercaptopyruvate sulfurtransferase (3-MST; Sigma, Taufkirchen, Germany), cystathionine-γ-lyase (CSE; Santa Cruz Biotechnology Inc., Heidelberg, Germany) or cystathionine-β-synthetase (CBS; Abnova, Heidelberg, Germany), hemeoxygenase-1 (HO-1; Biomol GmbH, Hamburg, Germany), heat shock protein 70 (HSP70; Abcam, Cambridge, UK), phosphorylated c-jun N-terminale kinase (pJNK), phosphorylated extracellular-signal regulated kinases (pERK1/2), phosphorylated p38 (pp38; all Cell Signaling, Frankfurt, Germany), NADPH oxidase 1 and 4 (Nox1; Nox4; Santa Cruz), or Nox2 (Becton Dickinson GmbH, Heidelberg, Germany). Normalization in order to control equal protein loading was performed by stripping and re-blotting of the membranes with glyceraldehyde-3-phosphate-dehydrogenase (GAPDH; Enzo Life Sciences GmbH, Lörrach, Germany) or β-tubulin (Cell Signaling). Data represent fold induction of indicated proteins with respect to GAPDH or β-tubulin after densitometric analysis.
Statistical Analysis
Experiments were performed with n = 6 mice per group. Power calculations were performed prior to the study in order to define group sizes. Graphs represent means ± standard error of means (SEM). Data were further analyzed for normal variation prior to one-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls post hoc test. In the case of failed normality testing, ANOVA on ranks (Kruskal-Wallis test) was used, followed by the Dunn’s post hoc test. P < 0.05 was considered significant. All calculations were performed with the SigmaPlot 11.0 statistical software (Systat Software Inc., Erkrath, Germany).
RESULTS
Effect of Hydrogen Sulfide on LPS-Induced Lung Damage and Inflammation
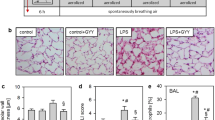
When compared to control animals in the presence or absence of H2S (Fig. 1a “control,” “control + H2S”), intranasal application of LPS led to a profound induction of histological lung damage after 6 h (Fig. 1a “LPS”), characterized by alveolar wall thickening and an increased ALI score (Fig. 1b, c). In contrast, inhalation of 80 ppm H2S for 6 h reduced these signs of histopathological damage despite LPS application (Fig. 1a–c). LPS treatment also induced neutrophil sequestration (Fig. 2a) and the release of the pro-inflammatory cytokine IL-1β (Fig. 2b) into the bronchoalveolar space compared to control or control + H2S mice. Likewise, H2S inhalation during LPS treatment reduced neutrophil influx and IL-1β release to control levels (Fig. 2).

Effect of hydrogen sulfide on LPS-induced lung damage. As controls, mice received phosphate-buffered saline (PBS, intranasally) and were kept in room air (control) or in 80 ppm H2S (control + H2S) for 6 h. LPS-treated mice (LPS i.n.) were either kept in room air (LPS) or in 80 ppm H2S (LPS + H2S) for 6 h. Sections from the left lung lobe were stained with hematoxylin and eosin. Representative pictures are shown for each experimental group (magnification = 200× (a)). High-power fields were randomly assigned to measure alveolar wall thickness (b) and to calculate an acute lung injury (ALI) score (c). Data represent means ± SEM for n = 6/group. ANOVA (Student-Newman-Keuls post hoc test), *P < 0.05 vs. control group; # P < 0.05 vs. control + H2S group; § P < 0.05 vs. LPS + H2S group.

Effect of hydrogen sulfide on LPS-induced lung inflammation. As controls, mice received phosphate-buffered saline (PBS, intranasally) and were kept in room air (control) or in 80 ppm H2S (control + H2S) for 6 h. LPS-treated mice (LPS i.n.) were either kept in room air (LPS) or in 80 ppm H2S (LPS + H2S) for 6 h. Bronchoalveolar lavage was performed in the right lung. The relative amount of neutrophils (a) was determined by cytospin analysis, and the amount of IL-1β (b) was determined by ELISA. Graphs represent means ± SEM, n = 6/group. ANOVA (Student-Newman-Keuls post hoc test) (a) or ANOVA on ranks (Dunn’s post hoc test) (b), *P < 0.05 vs. control group; # P < 0.05 vs. control + H2S group; § P < 0.05 vs. LPS + H2S group; n.d. not detectable.
Effect of Hydrogen Sulfide on CBS Protein Expression
We next sought to determine whether exogenous H2S-application exerts a feedback-loop on endogenous H2S production. Cystathionine-γ-lyase (CSE), cystathionine-β-synthetase (CBS), and 3-mercaptopyruvate sulfurtransferase (3-MST) catalyze H2S synthesis and are expressed in the lungs [13]. Compared to control animals, Western blot analysis of lung tissue revealed that expression of 3-MST and CSE was neither affected by LPS nor by H2S treatment or a combination of both (Fig. 3a, b). However, LPS instillation induced CBS expression which was absent in the presence of H2S inhalation (Fig. 3c, d).

Effect of hydrogen sulfide on CBS protein expression. As controls, mice received phosphate-buffered saline (PBS, intranasally) and were kept in room air (control) or in 80 ppm H2S (control + H2S) for 6 h. LPS-treated mice (LPS i.n.) were either kept in room air (LPS) or in 80 ppm H2S (LPS + H2S) for 6 h. Lung samples were taken from the upper right lobe for Western blot analysis. Normalization in order to control equal protein loading was performed by stripping and re-blotting of the membranes with glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) or β-tubulin. Representative Western blots are shown for 3-mercaptopyruvate sulfurtransferase (3-MST (a)), cystathionine-γ-lyase (CSE (b)), cystathionine-β-synthetase (CBS (c)), and β-tubulin (lower panel in a) and GAPDH (lower panel in b and c). Densitometric analysis of all samples were normalized to GAPDH and expressed as fold induction for CBS (d). Graphs represent means ± SEM, n = 6/group. ANOVA (Student-Newman-Keuls post hoc test), § P < 0.05 vs. LPS + H2S treated group.
Effect of Hydrogen Sulfide on Heat Shock Protein Expression
Other important pathways in the prevention of LPS-induced ALI are ascribed to the induction of stress protein signaling, e.g., hemoxygenase-1 (HO-1) and heat shock protein 70 (HSP70) [3]. HO-1 protein expression was not affected by LPS and/or additional H2S treatment (Fig. 4a). HSP70 protein expression was markedly reduced in lung tissue of LPS + H2S mice, compared to all other groups (Fig. 4b, c).

Effect of hydrogen sulfide on heat shock protein expression. As controls, mice received phosphate-buffered saline (PBS, intranasally) and were kept in room air (control) or in 80 ppm H2S (control + H2S) for 6 h. LPS-treated mice (LPS i.n.) were either kept in room air (LPS) or in 80 ppm H2S (LPS + H2S) for 6 h. Lung samples were taken from the upper right lobe for Western blot analysis. Normalization in order to control equal protein loading was performed by stripping and re-blotting of the membranes with glyceraldehyde-3-phosphate-dehydrogenase (GAPDH). Representative Western blots are shown for hemoxygenase-1 (HO-1 (a)) and heat shock protein 70 (Hsp70 (b)) and GAPDH (lower panel in a and b). Densitometric analysis of all samples were normalized to GAPDH and expressed as fold induction for Hsp70 (c). Graphs represent means ± SEM, n = 6/group. ANOVA (Student-Newman-Keuls post hoc test); § P < 0.05 vs. LPS + H2S group.
Effect of Hydrogen Sulfide on MAP Kinase Expression
Because regulation of MAP kinase signaling has been shown to confer protection against LPS-induced ALI [14], we evaluated lung tissue protein expression of phosphorylated JNK, ERK1/2, and p38 by Western blotting. Phosphorylation of JNK was slightly reduced in the LPS + H2S group compared to all other groups (Fig. 5a, b). In contrast to control and control + H2S animals, pERK1/2 protein expression was elevated after LPS treatment in the LPS and LPS + H2S group (Fig. 5c, d). However, statistical analysis of both pJNK and pERK1/2 did not reveal any differences. Expression of phosphorylated p38 MAPK (pp38 MAPK) was reduced by H2S inhalation, both in the presence and absence of LPS, while LPS alone had no influence on pp38 MAPK expression as compared to non-treated control animals (Fig. 5e, f).

Effect of hydrogen sulfide on MAP kinase expression. As controls, mice received phosphate-buffered saline (PBS, intranasally) and were kept in room air (control) or in 80 ppm H2S (control + H2S) for 6 h. LPS-treated mice (LPS i.n.) were either kept in room air (LPS) or in 80 ppm H2S (LPS + H2S) for 6 h. Lung samples were taken from the upper right lobe for Western blot analysis. Normalization in order to control equal protein loading was performed by stripping and re-blotting of the membranes with glyceraldehyde-3-phosphate-dehydrogenase (GAPDH). Representative Western blots are shown for pJNK (a), pERK1/2 (c), and pp38 (e) and GAPDH (lower panel in a, c, e). Densitometric analysis of all samples were normalized to GAPDH and expressed as fold induction for pJNK (b), pERK1/2 (d), and pp38 (f). Graphs represent means ± SEM, n = 6/group. ANOVA (Student-Newman-Keuls post hoc test); # P < 0.05 vs. control + H2S group; § P < 0.05 vs. LPS + H2S group.
Effect of Hydrogen Sulfide on Reactive Oxygen Species Production
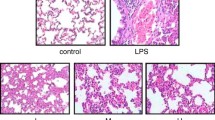
In LPS-induced ALI, the generation of ROS plays an essential role in the promotion of lung damage and inflammation [15]. We therefore evaluated, whether H2S affects ROS production in our model of pulmonary inflammation. DHE staining of lung tissue slices revealed that in contrast to control or control + H2S treated animals, LPS instillation alone caused significant ROS production (Fig. 6a “LPS”). This effect was prevented by additional H2S inhalation (Fig. 6a “LPS + H2S”). Quantification by densitometric analysis of all experimental groups and animals confirmed these results (Fig. 6b).

Effect of hydrogen sulfide on reactive oxygen species production. As controls, mice received phosphate-buffered saline (PBS, intranasally) and were kept in room air (control) or in 80 ppm H2S (control + H2S) for 6 h. LPS-treated mice (LPS i.n.) were either kept in room air (LPS) or in 80 ppm H2S (LPS + H2S) for 6 h. Sections from the right lung lobe were stained by dihydroethidium (DHE). Representative pictures are shown for each experimental group as indicated (magnification = 200× (a)). DHE fluorescence intensity was measured and expressed as fold induction compared to PBS + air group (e). Graphs represent means ± SEM, n = 6/group. ANOVA (Student-Newman-Keuls post hoc test); *P < 0.05 vs. control group; # P < 0.05 vs. control + H2S group; § P < 0.05 vs. LPS + H2S group.
Effect of Hydrogen Sulfide on NADPH Oxidases
Because the enzymatic generation of superoxide by NADPH oxidases (Nox) reflects the major source of ROS, we next investigated lung tissue protein expression of Nox1, Nox4, and Nox2, all previously described to be expressed in the lungs and to be involved in ROS mediated organ damage in several injury models [15]. Neither LPS nor H2S affected the expression of Nox1 or Nox4 (Fig. 7a, b). In contrast, LPS induced Nox2 as compared to the control. H2S inhalation, irrespective of LPS instillation, markedly reduced Nox2 protein expression in lung tissue (Fig. 7c, d).

Effect of hydrogen sulfide on NADPH oxidase expression. As controls, mice received phosphate-buffered saline (PBS, intranasally) and were kept in room air (control) or in 80 ppm H2S (control + H2S) for 6 h. LPS-treated mice (LPS i.n.) were either kept in room air (LPS) or in 80 ppm H2S (LPS + H2S) for 6 h. Lung samples were taken from the upper right lobe for Western blot analysis. Normalization in order to control equal protein loading was performed by stripping and re-blotting of the membranes with glyceraldehyde-3-phosphate-dehydrogenase (GAPDH). Representative Western blots are shown for Nox1 (a), Nox4 (b), Nox2 (c), and GAPDH (lower panel in a–c). Densitometric analysis of all samples were normalized to GAPDH and expressed as fold induction for Nox2 (d). Graphs represent means ± SEM, n = 6/group. ANOVA on ranks (Dunn’s post hoc test); # P < 0.05 vs. control + H2S group; § P < 0.05 vs. LPS + H2S group.
DISCUSSION
Acute lung injury resulting from lung infection remains a major problem in intensive care units and is associated with high morbidity and mortality rates. Despite modern antibiotics and supportive treatment, at present, specific therapeutic options in order to improve lung injury are lacking. In this respect, we have demonstrated recently that inhaled H2S in low dose can efficiently protect from ALI in a mouse model of pulmonary inflammation [11]. However, the underlying molecular pathways affected by H2S in this model remain elusive. In the present study, we show for the first time that H2S mediates this protection via the inhibition of LPS-induced inflammatory and oxidative processes, which in turn may result in the observed organ protection.
Hydrogen Sulfide Prevents Lung Damage by Inhibiting Inflammatory Processes
We have previously shown that inhalation of H2S averts the development of acute lung injury in a mouse model of LPS-induced acute lung injury [11]. In the current study, we again showed that LPS treatment clearly elevated histological signs of lung damage, e.g., alveolar wall thickening and ALI score as compared to controls. The effect was completely prevented by inhalation of H2S. Similar results have been obtained by us and others in mouse models of ventilator-induced lung injury [8, 16], hyperoxia-induced lung injury [9], or endotoxin-induced systemic inflammation [17], strongly underlining the lung protective capacity of H2S treatment.
Lung injury as a response to LPS instillation has been clearly linked to the elicited inflammatory response [18]. Neutrophil migration into lung tissue and the release of pro-inflammatory cytokines like IL-1β have been shown to mediate ALI [19]. In our model, LPS treatment led to a vast transmigration of neutrophils into the bronchoalveolar fluid that was accompanied by an increased IL-1β secretion. H2S inhalation on the other hand reduced both neutrophil recruitment and IL-1β release back to control levels. These findings are in line with recent publications demonstrating the anti-inflammatory properties of H2S in in vivo lung injury models like pulmonary inflammation [11, 17], mechanical ventilation [8], hyperoxia [9], or acute pancreatitis [20], reflecting that H2S-induced lung protection is mediated by anti-inflammatory effects.
Hydrogen Sulfide Inhibits CBS Expression in LPS-Induced ALI
In the present study, we determined the impact of LPS instillation with or without H2S inhalation on CSE, CBS, and 3-MST expression in the lung. These enzymes endogenously catalyze the synthesis of H2S [21] and are thought to be involved in organ protective effects in organ injury models [22,23,24,25]. Especially the regulation of CSE has been shown to exert organ protective effects in models of acute lung injury induced by acute pancreatitis [22, 24], but also all three were proven to be involved in attenuation of acute kidney injury in rats [23]. Our data reveal that lung protein expression of 3-MST and CSE was neither affected by LPS nor by H2S treatment. Although reduced CSE activity was attributed to LPS instillation in the rat lung [26, 27], we could not detect any effect on pulmonary CSE upon LPS challenge. In contrast, LPS instillation led to CBS induction that was prevented in the presence of H2S. Similar findings were reported in a recent publication of Wagner et al. [25]. Here, in a mouse model of blunt chest trauma, intravenous sulfide injection resulted in the attenuation of lung tissue apoptosis, which was accompanied by a reduced CBS and CSE protein expression [25]. Likewise, the application of exogenous H2S in our model may initiate the suppression of CBS activity, either as a result of reduced inflammation or as a direct negative feedback mechanism. Nonetheless, we show for the first time, that local administration of H2S regulates CBS expression in the lung, probably resulting in a reduction of endogenous H2S production.
Hydrogen Sulfide Prevents Heat Shock Protein Expression
Heat shock proteins like HO-1 or HSP70 are induced due to various insults in vivo and in vitro. Upregulation of both HO-1 and HSP70 in lung tissue have been described as a result of LPS stimulation and are clearly involved in protection against inflammation [3]. However, in our model, protein expression of HO-1 was unaffected by both LPS stimulation and/or additional H2S inhalation, while HSP70 expression was reduced due to the combination of LPS and H2S treatment. Although we did not find a role for HO-1 in our model, the results of HSP70 are in line with our recent observations in a model of ventilator-induced lung injury, where H2S treatment also clearly reduced HSP70 expression [8]. These results suggest that H2S may limit pro-inflammatory stress signaling in LPS-induced ALI.
Hydrogen Sulfide Prevents p38 MAP Kinase Signaling
Another possible pathway involved in the observed lung protective effects of H2S may be MAPK signaling. The regulation of MAPK activity has been reported to mediate inflammatory and/or oxidative effects upon infectious insults, thus augmenting lung injury [4, 28, 29]. Moreover, application of H2S has been described to block MAPK signaling [5, 6, 30]. Inhibition of p38 activation by H2S protected microglia from LPS-induced inflammation [5]. Both p38 and ERK 1/2 expression were reduced upon H2S application in PC-12 cells in a model of hypoxia-induced injury [6], and H2S prevented endothelial cell damage in rats in an ischemia-reperfusion rat model by blocking p38 and JNK signaling [30]. Our results showed no major effect of LPS or H2S on the regulation of pJNK or pERK1/2 expression. In contrast, phosphorylation of the p38 MAPK was significantly reduced by H2S inhalation, although LPS alone showed no effect. A more recent study reported that H2S treatment can inhibit p38 MAPK activation, leading to cellular protection in in vitro models of LPS-induced inflammation [5] or cobalt chloride-induced hypoxia [6]. Moreover, Sivarajah and co-workers demonstrated that the observed cardioprotective effects of H2S in a rat model of regional myocardial ischemia-reperfusion injury were at least in part due to a reduction in p38 phosphorylation [31], suggesting that H2S-related decreases in pp38 may play a role in protection from LPS-induced lung injury in our model.
Hydrogen Sulfide Prevents ROS Production and NADPH Oxidase 2 Signaling
It is widely accepted that pro-inflammatory signaling involves the accumulation of reactive oxygen species. Likewise, acute lung injury in response to LPS treatment is characterized by excessive ROS production [32] and its suppression has been shown to protect lungs from LPS-induced ALI in several models [15]. H2S can limit ROS either by directly scavenging free radicals, thus regulating oxidative signaling pathways, or by suppressing the inflammatory response [33]. Here, we clearly demonstrate that the vast ROS formation due to LPS challenge was prevented by H2S treatment. These findings are in line with recent studies carried out both by ourselves and others in vivo as well as in vitro [9, 34,35,36]. For instance, in mouse model of hyperoxia-induced, ROS were reduced by H2S application, which was accompanied by the inhibition of lung damage [9]. Likewise, in stimulated epithelial A549 cells [9], endothelial HUVEC cells [9], and RAW 264.7 macrophages [9], PC12 [35], and H9c2 [34] cells, application of H2S suppressed ROS formation and prevented the development of an inflammatory response.
With respect to an underlying mechanism, LPS-induced production of ROS is mainly attributed to the enzymatic activity of NADPH oxidases [37]. Several Nox family members have been described [32]. Among these, the regulation of Nox1, Nox2, and Nox4 were associated with acute lung injury [15, 38, 39]. In addition, H2S can reduce Nox expression in vivo and in vitro [40,41,42]. Our results show that neither LPS nor H2S influenced Nox1 or Nox4 regulation. In contrast, LPS-induced Nox2 expression was prevented in response to H2S inhalation. The importance of Nox2 regulation in lung oxidative stress and inflammation has been shown more recently [38, 39]. For instance, Menden and co-workers found in human pulmonary microvascular endothelial cells that LPS-induced oxidative stress and pro-inflammatory signaling was regulated by Nox2 [39]. In another LPS mouse model, Gandhirajan et al. showed that blockade of Nox2 signaling prevented vascular leakage and pulmonary edema in endothelial cells [38], suggesting that inhibition of Nox2 expression in our model, probably followed by downregulation of MAPK signaling, may also inhibit the subsequent development of ALI.
CONCLUSION
In the present study, inhalation of hydrogen sulfide prevents LPS-induced acute lung injury. CBS, HSP70, p38, Nox2 protein expression, and ROS production were all modulated due to H2S treatment. Although the results of the current study do not reveal the rate of contribution or a potential interaction between the signaling pathways, it appears reasonable that the anti-inflammatory effects of H2S-mediated lung protection are caused by limiting ROS production due to inhibition of Nox2 and p38 MAPK signaling pathways.
References
Martin, G.S., D.M. Mannino, S. Eaton, and M. Moss. 2003. The epidemiology of sepsis in the United States from 1979 through 2000. The New England Journal of Medicine 348: 1546–1554.
Matute-Bello, G., C.W. Frevert, and T.R. Martin. 2008. Animal models of acute lung injury. American Journal of Physiology. Lung Cellular and Molecular Physiology 295: L379–L399.
Wheeler, D.S., and H.R. Wong. 2007. Heat shock response and acute lung injury. Free Radical Biology & Medicine 42: 1–14.
Arndt, P.G., S.K. Young, and G.S. Worthen. 2005. Regulation of lipopolysaccharide-induced lung inflammation by plasminogen activator Inhibitor-1 through a JNK-mediated pathway. Journal of Immunology 175: 4049–4059.
Hu, L.F., P.T. Wong, P.K. Moore, and J.S. Bian. 2007. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. Journal of Neurochemistry 100: 1121–1128.
Lan, A., X. Liao, L. Mo, C. Yang, Z. Yang, X. Wang, F. Hu, P. Chen, J. Feng, D. Zheng, and L. Xiao. 2011. Hydrogen sulfide protects against chemical hypoxia-induced injury by inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells. PLoS One 6: e25921.
Biermann, J., W.A. Lagreze, N. Schallner, C.I. Schwer, and U. Goebel. 2011. Inhalative preconditioning with hydrogen sulfide attenuated apoptosis after retinal ischemia/reperfusion injury. Molecular Vision 17: 1275–1286.
Faller, S., S.W. Ryter, A.M. Choi, T. Loop, R. Schmidt, and A. Hoetzel. 2010. Inhaled hydrogen sulfide protects against ventilator-induced lung injury. Anesthesiology 113: 104–115.
Faller, S., S.G. Spassov, K.K. Zimmermann, S.W. Ryter, H. Buerkle, T. Loop, R. Schmidt, K.M. Strosing, and A. Hoetzel. 2013. Hydrogen sulfide prevents hyperoxia-induced lung injury by downregulating reactive oxygen species formation and angiopoietin-2 release. Current Pharmaceutical Design 19: 2715–2721.
Li, T., B. Zhao, C. Wang, H. Wang, Z. Liu, W. Li, H. Jin, C. Tang, and J. Du. 2008. Regulatory effects of hydrogen sulfide on IL-6, IL-8 and IL-10 levels in the plasma and pulmonary tissue of rats with acute lung injury. Exp Biol Med (Maywood ). 233: 1081–1087.
Faller, S., K.K. Zimmermann, K.M. Strosing, H. Engelstaedter, H. Buerkle, R. Schmidt, S.G. Spassov, and A. Hoetzel. 2012. Inhaled hydrogen sulfide protects against lipopolysaccharide-induced acute lung injury in mice. Medical Gas Research 2: 26.
Faller, S., K.M. Strosing, S.W. Ryter, H. Buerkle, T. Loop, R. Schmidt, and A. Hoetzel. 2012. The volatile anesthetic isoflurane prevents ventilator-induced lung injury via phosphoinositide 3-kinase/Akt signaling in mice. Anesthesia and Analgesia 114: 747–756.
Madden, J.A., S.B. Ahlf, M.W. Dantuma, K.R. Olson, and D.L. Roerig. 2012. Precursors and inhibitors of hydrogen sulfide synthesis affect acute hypoxic pulmonary vasoconstriction in the intact lung. Journal of Applied Physiology 112 (3): 411–418.
Zhang, T.Z., S.H. Yang, J.F. Yao, J. Du, and T.H. Yan. 2015. Sangxingtang inhibits the inflammation of LPS-induced acute lung injury in mice by down-regulating the MAPK/NF-kappaB pathway. Chinese Journal of Natural Medicines 13: 889–895.
Carnesecchi, S., J.C. Pache, and C. Barazzone-Argiroffo. 2012. NOX enzymes: potential target for the treatment of acute lung injury. Cellular and Molecular Life Sciences 69: 2373–2385.
Francis, R.C., K. Vaporidi, K.D. Bloch, F. Ichinose, and W.M. Zapol. 2011. Protective and detrimental effects of sodium sulfide and hydrogen sulfide in murine ventilator-induced lung injury. Anesthesiology 115: 1012–1021.
Tokuda, K., K. Kida, E. Marutani, E. Crimi, M. Bougaki, A. Khatri, H. Kimura, and F. Ichinose. 2012. Inhaled hydrogen sulfide prevents endotoxin-induced systemic inflammation and improves survival by altering sulfide metabolism in mice. Antioxidants & Redox Signaling 17: 11–21.
Hasan, B., F.S. Li, A. Siyit, E. Tuyghun, J.H. Luo, H. Upur, and A. Ablimit. 2014. Expression of aquaporins in the lungs of mice with acute injury caused by LPS treatment. Respiratory Physiology & Neurobiology 200C: 40–45.
Li, L., G. Rossoni, A. Sparatore, L.C. Lee, P. Del Soldato, and P.K. Moore. 2007. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radical Biology & Medicine 42: 706–719.
Sidhapuriwala, J.N., S.W. Ng, and M. Bhatia. 2009. Effects of hydrogen sulfide on inflammation in caerulein-induced acute pancreatitis. Journal Inflammation (London) 6: 35.
Huang, C.W., and P.K. Moore. 2015. H2S synthesizing enzymes: biochemistry and molecular aspects. Handbook of Experimental Pharmacology 230: 3–25.
Ang, A.D., J. Rivers-Auty, A. Hegde, I. Ishii, and M. Bhatia. 2013. The effect of CSE gene deletion in caerulein-induced acute pancreatitis in the mouse. American Journal of Physiology. Gastrointestinal and Liver Physiology 305: G712–G721.
Dugbartey, G.J., F. Talaei, M.C. Houwertjes, M. Goris, A.H. Epema, H.R. Bouma, and R.H. Henning. 2015. Dopamine treatment attenuates acute kidney injury in a rat model of deep hypothermia and rewarming—the role of renal H2S-producing enzymes. European Journal of Pharmacology 769: 225–233.
Qu, Z., Y. Jiang, B.Q. Wu, Y.F. Duan, Z.D. Sun, and G.H. Luo. 2014. Cystathionine-gamma-lyase inhibitor attenuates acute lung injury induced by acute pancreatitis in rats. Archives of Medical Science 10: 825–829.
Wagner, F., A. Scheuerle, S. Weber, B. Stahl, O. McCook, M.W. Knoferl, M. Huber-Lang, D.H. Seitz, J. Thomas, P. Asfar, C. Szabo, P. Moller, F. Gebhard, M. Georgieff, E. Calzia, P. Radermacher, and K. Wagner. 2011. Cardiopulmonary, histologic, and inflammatory effects of intravenous Na2S after blunt chest trauma-induced lung contusion in mice. The Journal of Trauma 71: 1659–1667.
Zhou, X.H., P. Wei, X.L. Huang, and Y.L. Ling. 2009. Role of endogenous and exogenous hydrogen sulfide in acute lung injury induced by LPS in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi 25: 289–294.
Zhou, X.H., X.L. Huang, P. Wei, F.J. Tian, and Y.L. Ling. 2009. Role of hydrogen sulfide/cystathionine-gamma-lyase system in acute lung injury induced by lipopolysaccharide in rats. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 21: 199–202.
Asaduzzaman, M., Y. Wang, and H. Thorlacius. 2008. Critical role of p38 mitogen-activated protein kinase signaling in septic lung injury. Critical Care Medicine 36: 482–488.
Schuh, K., and A. Pahl. 2009. Inhibition of the MAP kinase ERK protects from lipopolysaccharide-induced lung injury. Biochemical Pharmacology 77: 1827–1834.
Issa, K., A. Kimmoun, S. Collin, F. Ganster, S. Fremont-Orlowski, P. Asfar, P.M. Mertes, and B. Levy. 2013. Compared effects of inhibition and exogenous administration of hydrogen sulphide in ischaemia-reperfusion injury. Critical Care 17: R129.
Sivarajah, A., M. Collino, M. Yasin, E. Benetti, M. Gallicchio, E. Mazzon, S. Cuzzocrea, R. Fantozzi, and C. Thiemermann. 2009. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 31: 267–274.
Lee, I.T., and C.M. Yang. 2012. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochemical Pharmacology 84: 581–590.
Stein, A., and S.M. Bailey. 2013. Redox biology of hydrogen sulfide: implications for physiology, pathophysiology, and pharmacology. Redox Biology 1: 32–39.
Dong, X.B., C.T. Yang, D.D. Zheng, L.Q. Mo, X.Y. Wang, A.P. Lan, F. Hu, P.X. Chen, J.Q. Feng, M.F. Zhang, and X.X. Liao. 2012. Inhibition of ROS-activated ERK1/2 pathway contributes to the protection of H2S against chemical hypoxia-induced injury in H9c2 cells. Molecular and Cellular Biochemistry 362: 149–157.
Lan, A., W. Xu, H. Zhang, X. Hua, D. Zheng, R. Guo, N. Shen, F. Hu, J. Feng, and D. Liu. 2013. Inhibition of ROS-activated p38MAPK pathway is involved in the protective effect of H2S against chemical hypoxia-induced inflammation in PC12 cells. Neurochemical Research 38: 1454–1466.
Yu, Q., Z. Lu, L. Tao, L. Yang, Y. Guo, Y. Yang, X. Sun, and Q. Ding. 2015. ROS-dependent neuroprotective effects of NaHS in ischemia brain injury involves the PARP/AIF pathway. Cellular Physiology and Biochemistry 36: 1539–1551.
Kratzer, E., Y. Tian, N. Sarich, T. Wu, A. Meliton, A. Leff, and A.A. Birukova. 2012. Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. American Journal of Respiratory Cell and Molecular Biology 47: 688–697.
Gandhirajan, R.K., S. Meng, H.C. Chandramoorthy, K. Mallilankaraman, S. Mancarella, H. Gao, R. Razmpour, X.F. Yang, S.R. Houser, J. Chen, W.J. Koch, H. Wang, J. Soboloff, D.L. Gill, and M. Madesh. 2013. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. The Journal of Clinical Investigation 123: 887–902.
Menden, H., E. Tate, N. Hogg, and V. Sampath. 2013. LPS-mediated endothelial activation in pulmonary endothelial cells: role of Nox2-dependent IKK-beta phosphorylation. American Journal of Physiology. Lung Cellular and Molecular Physiology 304: L445–L455.
Li, H.D., Z.R. Zhang, Q.X. Zhang, Z.C. Qin, D.M. He, and J.S. Chen. 2013. Treatment with exogenous hydrogen sulfide attenuates hyperoxia-induced acute lung injury in mice. European Journal of Applied Physiology 113: 1555–1563.
Tyagi, N., K.S. Moshal, U. Sen, T.P. Vacek, M. Kumar, W.M. Hughes Jr., S. Kundu, and S.C. Tyagi. 2009. H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxidants & Redox Signaling 11: 25–33.
Zheng, D., S. Dong, T. Li, F. Yang, X. Yu, J. Wu, X. Zhong, Y. Zhao, L. Wang, C. Xu, F. Lu, and W. Zhang. 2015. Exogenous hydrogen sulfide attenuates cardiac fibrosis through reactive oxygen species signal pathways in experimental diabetes mellitus models. Cellular Physiology and Biochemistry 36: 917–929.
Acknowledgments
This study was supported by a grant from the Deutsche Forschungsgemeinschaft (Bonn, Germany) to Alexander Hoetzel (DFG HO 2464/3-1).
The authors thank E. Bodurova (Life Imaging Centre, Albert-Ludwigs-University Freiburg, Germany) for the excellent technical assistance.
Parts of the study have been published online as the doctoral thesis by Kornelia K. Zimmermann, 2013: “Die protektive Wirkung Schwefelwasserstoffs bei oxidativer und inflammatorischer Lungenschädigung” [German] https://www.freidok.uni-freiburg.de/dnb/download/9093.
Author information
Authors and Affiliations
Contributions
KKZ helped to conduct the study and analyzed the data; KMS helped to conduct the study; PMI helped to design and conduct the study and to analyze the data; HE helped to analyze the data and critically revised the manuscript; SGS helped to conduct the study and analyzed the data; AH helped to design and conduct the study, to analyze the data and to write the manuscript; and SF designed and conducted the study, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
This study was supported by a grant from the Deutsche Forschungsgemeinschaft (Bonn, Germany) to Alexander Hoetzel (DFG HO 2464/3-1). All other authors declare that they have no competing interests.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted (Ethics Committee University of Freiburg and Regierungspräsidium Freiburg, Freiburg, Germany, Permission No. G-07/25).
Rights and permissions
About this article

Cite this article
Zimmermann, K.K., Spassov, S.G., Strosing, K.M. et al. Hydrogen Sulfide Exerts Anti-oxidative and Anti-inflammatory Effects in Acute Lung Injury. Inflammation 41, 249–259 (2018). https://doi.org/10.1007/s10753-017-0684-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-017-0684-4