
Abstract
V122I genotype variant (pV142I) is the most common hereditary transthyretin amyloidosis (hATTR) in the USA, with 3–3.5% of African-Americans being the carriers of this mutation. We aimed to compare baseline clinical features, cardiac parameters, and mortality in V122I-ATTR with the wild-type ATTR and other hATTR subtypes. We systematically searched PubMed/Medline and Google Scholar databases to identify relevant studies from inception to 10th September, 2020 reporting phenotypic, echocardiographic, and/or laboratory parameters in patients with hereditary and wild types of cardiac amyloidoses. A total of 2843 patients from 7 individual studies with 67–100% males and an overall follow-up duration of 51.6 ± 30.4 months were identified. The mean age of diagnosis among wild-type ATTR patients was 77 years, followed by 71.2 and 65 years in V122I and T60A group patients, respectively. V122I patients were mostly black, had a poor quality of life, and highest mortality risk compared with other subtypes. Merely, the presence of V122I mutation was identified as an independent predictor of mortality. V30M subtype correlated with the least severe cardiac disease and a median survival duration comparable with T60A subtype. V122I ATTR is an aggressive disease, prevalent in African-Americans, and is associated with a greater morbidity and mortality, which is partly attributed to its misdiagnosis and/or late diagnosis. Current advances in non-invasive studies to diagnose hATTR coupled with concurrent drug therapies have improved quality of life and provide a survival benefit to these patients.

Similar content being viewed by others

References
Falk RH, Comenzo RL, Skinner M (1997) The systemic amyloidoses. N Engl J Med 337(13):898–909. https://doi.org/10.1056/NEJM199709253371306
Kelly JW, Colon W, Lai Z, Lashuel HA, McCulloch J, McCutchen SL, Miroy GJ, Peterson SA (1997) Transthyretin quaternary and tertiary structural changes facilitate misassembly into amyloid. Adv Protein Chem 50:161–181. https://doi.org/10.1016/s0065-3233(08)60321-6
Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS (2019) Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol 73(22):2872–2891. https://doi.org/10.1016/j.jacc.2019.04.003
Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, Steidley DE, Ventura H, Murali S, Silver MA, Jacoby D, Fedson S, Hummel SL, Kristen AV, Damy T, Plante-Bordeneuve V, Coelho T, Mundayat R, Suhr OB, Waddington Cruz M, Rapezzi C, Investigators T (2016) Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol 68(2):161–172. https://doi.org/10.1016/j.jacc.2016.03.596
Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, Berk JL, Plante-Bordeneuve V, Schmidt HHJ, Merlini G (2015) Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol 66(21):2451–2466. https://doi.org/10.1016/j.jacc.2015.09.075
Grogan M, Hawkins PN, Kristen AV, Berk JL, Suhr OB, Lin H, Merkel M, McManus A, Powell C, Vest J, Karsten V, Judge DP (2019) Identifying mixed phenotype: evaluating the presence of polyneuropathy in patients with hereditary transthyretin-mediated amyloidosis with cardiomyopathy. J Card Fail 25(8):S09-S10. https://doi.org/10.1016/j.cardfail.2019.07.031
Castano A, Drachman BM, Judge D, Maurer MS (2015) Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev 20(2):163–178. https://doi.org/10.1007/s10741-014-9462-7
Givens RC, Russo C, Green P, Maurer MS (2013) Comparison of cardiac amyloidosis due to wild-type and V122I transthyretin in older adults referred to an academic medical center. Aging health 9(2):229–235. https://doi.org/10.2217/ahe.13.10
Dungu JN, Papadopoulou SA, Wykes K, Mahmood I, Marshall J, Valencia O, Fontana M, Whelan CJ, Gillmore JD, Hawkins PN, Anderson LJ (2016) Afro-Caribbean heart failure in the United Kingdom: cause, outcomes, and ATTR V122I cardiac amyloidosis. Circ Heart Fail 9(9):e003352. https://doi.org/10.1161/CIRCHEARTFAILURE.116.003352
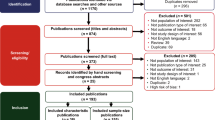
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 6(7):e1000097. https://doi.org/10.1371/journal.pmed.1000097
Biomedicine IoMUCotSaEIoDi (1995) Society's choices: social and ethical decision making in biomedicine. National Academic Press (US), Washington (DC). https://doi.org/10.17226/4771
Slim K, Nini E, Forestier D, Kwiatkowski F, Panis Y, Chipponi J (2003) Methodological index for non-randomized studies (minors): development and validation of a new instrument. ANZ J Surg 73(9):712–716. https://doi.org/10.1046/j.1445-2197.2003.02748.x
Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, Falk RH, Cheung KN, Patel AR, Pano A, Packman J, Grogan DR (2012) Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the transthyretin amyloidosis cardiac study (TRACS). Am Heart J 164 (2):222–228 e221. https://doi.org/10.1016/j.ahj.2012.04.015
Arruda-Olson AM, Zeldenrust SR, Dispenzieri A, Gertz MA, Miller FA, Bielinski SJ, Klarich KW, Scott CG, Grogan M (2013) Genotype, echocardiography, and survival in familial transthyretin amyloidosis. Amyloid 20(4):263–268. https://doi.org/10.3109/13506129.2013.845745
Swiecicki PL, Zhen DB, Mauermann ML, Kyle RA, Zeldenrust SR, Grogan M, Dispenzieri A, Gertz MA (2015) Hereditary ATTR amyloidosis: a single-institution experience with 266 patients. Amyloid 22(2):123–131. https://doi.org/10.3109/13506129.2015.1019610
Chacko L, Martone R, Bandera F, Lane T, Martinez-Naharro A, Boldrini M, Rezk T, Whelan C, Quarta C, Rowczenio D, Gilbertson JA, Wongwarawipat T, Lachmann H, Wechalekar A, Sachchithanantham S, Mahmood S, Marcucci R, Knight D, Hutt D, Moon J, Petrie A, Cappelli F, Guazzi M, Hawkins PN, Gillmore JD, Fontana M (2020) Echocardiographic phenotype and prognosis in transthyretin cardiac amyloidosis. Eur Heart J 41(14):1439–1447. https://doi.org/10.1093/eurheartj/ehz905
Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, Rowczenio DM, Gilbertson JA, Hutt DF, Rezk T, Strehina SG, Caringal-Galima J, Manwani R, Sharpley FA, Wechalekar AD, Lachmann HJ, Mahmood S, Sachchithanantham S, Drage EPS, Jenner HD, McDonald R, Bertolli O, Calleja A, Hawkins PN, Gillmore JD (2019) Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation 140(1):16–26. https://doi.org/10.1161/CIRCULATIONAHA.118.038169
Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN (1997) Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in Black Americans. N Engl J Med 336(7):466–473. https://doi.org/10.1056/NEJM199702133360703
Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C, Investigators A-AS (2018) Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379(11):1007–1016. https://doi.org/10.1056/NEJMoa1805689
Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Plante-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH 3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB (2018) Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379(1):11–21. https://doi.org/10.1056/NEJMoa1716153
Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Plante-Bordeneuve V, Barroso FA, Merlini G, Obici L, Scheinberg M, Brannagan TH 3rd, Litchy WJ, Whelan C, Drachman BM, Adams D, Heitner SB, Conceicao I, Schmidt HH, Vita G, Campistol JM, Gamez J, Gorevic PD, Gane E, Shah AM, Solomon SD, Monia BP, Hughes SG, Kwoh TJ, McEvoy BW, Jung SW, Baker BF, Ackermann EJ, Gertz MA, Coelho T (2018) Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 379(1):22–31. https://doi.org/10.1056/NEJMoa1716793
Acknowledgements
None.
Author information
Authors and Affiliations
Contributions
AG, SL, and ZS designed the study concept. AG, SL, and TD wrote the original draft and designed the schematic tables. SR, VBB, and RRP edited tables and revised the manuscript. AG, TD, and ZS performed the final revisions and approved the final version of the article after reviewing feedback from all other authors and reviewers. All authors contributed to study design, critically reviewed the first draft, approved the final version, and agreed to be accountable for the work.
Corresponding author
Ethics declarations
Ethical approval
Our study, being a systematic review of literature, did not require an approval from the institutional review board.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Goyal, A., Lahan, S., Dalia, T. et al. Clinical comparison of V122I genotypic variant of transthyretin amyloid cardiomyopathy with wild-type and other hereditary variants: a systematic review. Heart Fail Rev 27, 849–856 (2022). https://doi.org/10.1007/s10741-021-10098-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-021-10098-6