
Abstract
Bacterial wilt, caused by Ralstonia solanacearum, is a devastating disease resulting in tremendous losses of economic crops such as plants in the Solanaceae. Recent studies showed that R. solanacearum is spreading from the lowlands to the highlands in China. We studied 97 Chinese R. solanacearum strains that were isolated from four tobacco-growing zones over a wide range of elevations using phylotype specific multiplex polymerase chain reaction (Pmx-PCR) and phylogenetic relationships (egl and mutS). The results showed that all isolates belonged to phylotype I, which were further clustered into eight egl-sequence type groups (egl-group, sequevar): sequevars 13, 14, 15, 17, 34, 44, 54, and 55. In addition, Sequevar 55, found from the highlands, was a new/unknown one. Southeast China (Z3) had the largest number of egl-groups, containing six sequevars. The basin of the Yangzi River (Z1) and southwestern China (Z2) contained five egl-groups. The basin of the Huai River (Z4), near the north of China, where slight bacterial wilt occurred recently, contained a single group, sequevar 15. The distribution of sequevars was associated with elevation. Sequevar 15 was over-represented in lowland elevations, while sequevar 54 and the new/unknown one were only found in areas of moderate to high elevations. This finding suggested that the phylotype I strains infecting tobacco were diverse in China and regional integrated control strategies should be considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ralstonia solanacearum, widely distributed in tropical, subtropical, and temperate regions of the world, is a complex species with considerable diversity (Hayward 1991; Genin and Denny 2012). A large number of studies have focused on the global genetic diversity of the pathogen. Early attempts have subdivided the R. solanacearum strains into five races and six biovars. Based on the host range, R. solanacearum has been phenotypically classified into five races: Race 1 (solanaceous vegetables), Race 2 (banana), Race 3 (potato and tomato from temperate regions), Race 4 (ginger), and Race 5 (mulberry) (Buddenhagen et al. 1962; Wicker et al. 2007). Biovar classification has been defined by their ability to utilize disaccharides and to oxidize hexose alcohols (He et al. 1983; Hayward 1964, 1991; Genin and Denny 2012). Although both classification schemes have been used to describe the diversity of R. solanacearum for a long time, several disadvantages have been revealed with our increasing knowledge of pathogen diversity, such as the lack of discriminating power, additional time required, and lack of correlation with the phylogenetic origin of different strains (Siri et al. 2011; Genin and Denny 2012).
To improve our understanding of the genetic relationship among the R. solanacearum species complex, DNA-based analyses were used, such as restriction fragment length polymorphisms (RFLP), polymerase chain reaction (PCR) -RFLP, 16S rRNA gene sequence analysis, repetitive extragenic palindromic (REP)-PCR, and amplified fragment length polymorphism (AFLP) (Cook et al. 1989; Li et al. 1993; Frey et al. 1996; Poussier et al. 2000b; Horita and Tsuchiya 2001). As examination of the diversity of the strains increased, it became clear that the R. solanacearum strains had four major phylotype subdivisions (Fegan and Prior 2005). Phylotype can be rapidly assigned by a multiplex PCR (Pmx-PCR). Each phylotype is subdivided into numbered sequevars or closely related individual strains that are usually identified by analyzing the sequence similarity of the endoglucanase gene (egl, a conserved virulence factor) (Fegan and Prior 2005). The nucleotide sequences of mutS (a DNA mismatch repair enzyme), hrpB (a regulator of type III secretion) and pga (polygalacturonase precursor gene) have also been used for the subdivision of the species complex (Prior and Fegan 2005; Ramesh et al. 2014). The phylotypes correspond closely to the geographical origins of the strains: Phylotype I includes strains from Asia; phylotype II, strains from the Americas; phylotype III, strains from Africa and surrounding islands; and phylotype IV, strains isolated primarily from Indonesia, as well as two closely related species, R. syzygii and banana blood disease bacterium (BDB). Phylotype II has two clearly recognizable sub-clusters, phylotype IIA and phylotype IIB (Prior and Fegan 2005; Fegan and Prior 2006; Castillo and Greenberg 2007).
Bacterial wilt is one of the most important diseases in China. There are many reports studying the genetic diversity of Chinese R. solanacearum strains; three out of five races (race 1, 3 and 5) and all five biovars (bv. 1, 2, 3, 4 and 5) have been identified (He et al. 1983; Lu 1998). Xu et al. (2009) analyzed the genetic diversity of Chinese R. solanacearum strains, suggesting that Chinese R. solanacearum strains belonged to phylotype I and II and that phylotype I was predominant and included 10 different sequevars. According to Xue et al. (2011), the majority of tested Chinese strains belonged to Asian phylotype I, which had a high diversity from the BOX-PCR fingerprints generated from the genomic DNA. In Taiwan, R. solanacearum strains mainly belonged to phylotype I and sequevar 15 was predominant (Lin et al. 2014). To date, very limited success has been achieved for the control of bacterial wilt caused by R. solanacearum. The high diversity of the pathogen may be one of the reasons.
Recently, some reports showed that R. solanacearum is spreading from the lowlands to the highlands and cold areas in China as a result of the changed climate, ecological environment, and planting structure adjustment (Kong 2003; Zhou et al. 2012). Tobacco is one of the most important economic crops in China, and tobacco planting areas are widely distributed in mountain, hill, and lowland regions (Hayward 1991; Zhu et al. 2002). Based on this information, Chinese R. solanacearum strains isolated from Nicotiana tabacum are suitable candidates for studying the relationship between the elevation and genetic structures of R. solanacearum. Li et al. (2016) have reported the genetic diversity of Chinese tobacco R. solanacearum, but they did not explain the relationship between genetic structure and elevation. The aims of this study were to analyze: i) genetic diversity of R. solanacearum strains collected from all tobacco-growing zones in China, and ii) whether elevation affects phylogenetic diversity in China.
Materials and methods
Bacterial isolates and growth conditions
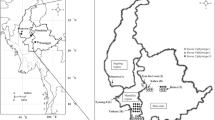
A total of 97 R. solanacearum isolates were isolated from wilting tobacco plants from all tobacco-growing areas that are clustered in four tobacco-growing zones from 2013 to 2015 in China (Table 1, Supplemental Table 1). In total, 56 counties from 13 provinces were include: Z1, the basin of the Yangzi River, contained five provinces (Chongqing, Guizhou, Shanxi, Hubei, and Hunan); Z2, southwestern China, contained three provinces (Sichuan, Yunnan, and Guangxi); Z3, southeast China, contained three provinces (Jiangxi, Guangdong, and Fujian); and Z4, the basin of the Huai River, contained three provinces (Anhui, Henan, and Shandong). According to the elevations, all areas were classified into three regions: region 1 (R1, highlands) with elevations over 1500 m a.s.l.; region 2 (R2, the areas of moderate elevations) with elevations from 500 m a.s.l. to 1500 m a.s.l.; and region 3 (R3, lowlands) with elevations below 500 m a.s.l. (Table 2).
For the isolation of R. solanacearum, infected stem tissues were first surface disinfected with 75 % ethanol for 3 min, peeled, subsampled and macerated in sterile distilled water for 30 min. Macerates were then streaked on Kelman’s triphenyltetrazolium chloride (TZC) agar medium (Kelman 1954). Plates were incubated at 30 ± 2 °C for 48 h. Bacterial colonies displaying the typical irregular, mucoid, bird’s eye aspect were then streaked onto fresh TZC medium for further purification. All isolated strains were named and maintained in 20 % glycerol at −80 °C.
DNA assay and phylotype identification
The total DNA from each strain was prepared using TIANamp Bacterial DNA Kit (Tiangen Biotech, Beijing, China), according to the manufacturer’s instructions. The phylotype identity of each strain was determined by multiplex PCR as described by Fegan and Prior (2005). Pmx-PCR was carried out in 25 μl of reaction mixture containing 1× Taq MasterMix (PCR buffer, 1.5 mM MgCl2, 250 μM of each dNTP, 50 mM KCl, 10 mM Tris-HCl and 1.25 U of Taq DNA polymerase) (Tiangen Biotech), 6 pmoles of primers Nmult:21:1F, Nmult:21:2F, Nmult:22:InF, 18 pmoles of the primer Nmult:23:AF and 4 pmoles of primers 759 and 760 (all primers are listed in Supplemental Table 2) (Opina et al. 1997; Xu et al. 2009). This Pmx-PCR amplifies a 281-bp “universal” R. solanacearum specific reference band in addition to the following phylotype-specific PCR products: a 144-bp band from phylotype I strains; a 372-bp band from phylotype II strains; a 91-bp amplicon from phylotype III strains; and a 213-bp band from phylotype IV strains. The PCR program was carried out in a thermocycler as previously described (Xu et al. 2009). Then, 5 μl of PCR product was subjected to electrophoresis on 1.5 % agarose gel, stained with ethidium bromide and visualized on a UV-transilluminator.
Sequencing of egl and mutS genes
The phylogeny of each strain was characterized using comparative analysis of the partial nucleotide sequences of egl and mutS genes. Primers were used for amplifying the partial egl and mutS genes as previously described (Poussier et al. 2000a; Prior and Fegan 2005). Fifty microliters of the reaction mixture contained 1× Taq MasterMix (Tiangen Biotech), 0.25 μM of each primer, and 50 ng of DNA as the template (Xu et al. 2009). The PCR programs of the egl and mutS genes were performed as previously described (Prior and Fegan 2005; Xu et al. 2009). Fifty microliters of reaction mixtures were examined by electrophoresis on 1.5 % agarose gels in TAE buffer. Bands were revealed by visualization with UV light after ethidium bromide staining. PCR products were sequenced by the Beijing genomics institute. The primers mentioned above were also used as sequencing primers.
Phylogenetic analysis
Thirty-four reference strain sequences were selected and used in the analysis (Supplemental Table 3). Sequences were manually edited and trimmed using BioEdit 7.0.5.1 (Hall 1999), then aligned using Clustal x (Jeanmougin et al. 1998). Phylogenetic analysis was performed using neighbor-joining (NJ) and the algorithm of Jukes and Cantor with 1000 bootstrap resamplings in MEGA version 5 (Tamura et al. 2011) along with reference sequences from NCBI. Sequence extremities were trimmed on the basis of sequence quality and reading frame as previously described (Deberdt et al. 2014; N’Guessan et al. 2012; Wicker et al. 2012), giving final egl sequences of 666 nucleotides and mutS of 651 nucleotides. All egl and mutS sequences from the R. solanacearum strains used in this study were deposited into the GenBank (accession numbers of egl: KT961427 - KT961523, accession numbers of mutS: KT961524 - KT961620).
Pathogenicity test
A tobacco susceptible cultivar (cv. Yunyan 87) was used for the pathogenicity test. Two-week-old seedlings were transplanted in plastic pots (9 cm in diameter) containing horticultural soil and then grown in a greenhouse at 30 °C and 25 °C day and night temperatures and a 14- and 10-h photoperiod, respectively. Plants with four to five true leaves were inoculated by soil drenching. Briefly, the strain was grown on B medium (Boucher et al. 1985) for 16 h and then suspended in sterile deionized water. Each plant was inoculated by pouring 10 mL of suspension (OD600 = 0.1, approximately 1.0 × 108 cells ml−1). The incidence and disease index were monitored every day for 28 days using a 0 to 4 scale: 0 = asymptomatic plant, 1 = less than 25 % wilted leaves, 2 = less than 50 % wilted leaves, 3 = more than 50 % wilted leaves, and 4 = completely wilted leaves (dead plant) (Lebeau et al. 2013). The experiment was repeated twice. Analysis of variance (ANOVA) was used to analyse the disease index data at each assessment date using Statistical Product and Service Solutions (SPSS).
Results
R. solanacearum strain collection, identification and phylotype analysis
A total of 97 R. solanacearum isolates collected from four tobacco-growing zones were isolated and identified (Supplemental Table 1). There were 44 isolates in Z1, 21 in Z2, 27 in Z3, and only five in Z4. On TZC agar medium, these strains yielded typical pink virulent type colonies that were cream colored, irregularly shaped, and highly fluidal. All of these isolates were further confirmed by PCR using the R. solanacearum specific universal primer 759/760, which gave a specific 281-bp band. Using the Pmx-PCR protocol with primers designed from the ITS region, the 144-bp fragment specific to phylotype I and the 281-bp fragment universal to R. solanacearum species complex were observed with all tested isolates, suggesting that all of the isolates in this study were phylotype I strains.
egl and mutS gene analysis
Partial nucleotide sequences of the egl gene and the mutS gene from 97 R. solanacearum isolates were aligned. The phylogenetic relationships according to egl gene sequences were analyzed together with 34 reference strains and mutS genes with 29 reference strains. Sequences of reference strains were added to the trees in order to position the tested strains within the known phylogenetic structure. The phylogenetic trees based on egl and mutS partial sequences were in global agreement (Figs. 1 and 2).

Phylogenetic neighbor-joining tree based on partial endoglucanase (egl) gene sequences of Ralstonia solanacearum strains from China and reference strains. Values at the branches indicate the percentage of bootstrap support for 1000 resamplings. Strains with the exact same sequence were reduced to a single representative taxon for analysis, but individual names are all displayed on the tree. Black circles indicate reference strains

Phylogenetic neighbor-joining tree based on the partial DNA mismatch repair protein (mutS) gene sequences of Ralstonia solanacearum strains from China and reference strains. Values at the branches indicate the percentage of bootstrap support for 1000 resamplings. Strains with the exact same sequence were reduced to a single representative taxon for analysis, but individual names are all displayed on the tree. Black circles indicate reference strains
Based on the egl-tree (Fig. 1), all isolates from this study were classified into phylotype I, which was consistent with the Pmx-PCR. The egl-tree branching pattern within phylotype I showed a split with a 98 % bootstrap value. According to the sequevar definition and the position of reference strains in the egl-tree, all tested strains were further clustered into seven major groups that were defined as egl-sequence type groups (egl-group) A, B, C, D, E, F, and G, representing sequevars 13, 14, 15, 17, 34, 44, and 54, respectively. In addition, YNLXBS-1, collected from a highland elevation (1838 m) (Supplemental Table 1), was not identical to any previously designated sequevar, and was defined as sequevar 55 (Fig. 1). According to the mutS-tree, there were two mutS-based sequence types (mutS-ST A and B) (Fig. 2).
Distribution of sequevars in different zones
Sequevars were determined from the egl sequences. The most widespread one was sequevar 17 (group D) (26.80 % of the tested strains; Table 3), containing reference strain P11 (Xu et al. 2009), which was found in three tobacco-growing zones (Z1, Z2, and Z3). The second one was sequevar 15 (group C), with reference strain PSS358 (Lin et al. 2014), which was found in Z1, Z3, and Z4. In Z4 (the Basin of the Huai River), sequevar 15 was the only observed sequevar. Sequevar 44 (group F), with reference strain Bd11 (Xu et al. 2009), was distributed in Z1, Z2, and Z3. Sequevar 54 (group G), containing reference strain HBJS1 (Li et al. 2016), was identified in Z1 and Z2. Sequevar 34 (group E), with reference strain PSS219 (Xu et al. 2009), was found in Z1 and Z3. Sequevar 13 (group A), with reference strain JT523 (Poussier et al. 2000a), was identified in Z2 and Z3. Sequevar 14 (group B) contained three isolates and was only found in Z3. Sequevar 55 (only one strain, YNLXBS-1), was collected from Z2. The distribution of sequevars significantly differs across tobacco-growing zones (χ2 test, P = 5.10× 10−8).
Relationship between the elevation and sequevar composition
Sequevars 17 and 44 were prevalent over a wide range of elevations. Sequevars 15 and 34 were clearly overrepresented in lowland elevations. Conversely, sequevar 54 and 55 (group G and the unknown YNLXBS-1) were only found in moderate and high elevations (Fig. 3).

Box chart of elevation distribution of every sequevar. Sequevars (groups) were based on phylogenetic neighbor-joining tree of egl gene sequences
To investigate the possible impact of elevation on sequevar composition, we clustered all areas into three regions according to the elevation ranges (R1, R2, and R3) (Table 2). The distribution of egl-ST groups (Table 4) was significantly different between R1, R2, and R3 (χ2 test, P = 5.55× 10−8). Sequevar 15 was significantly overrepresented in lowland elevations (R3) (χ2 test, P = 1.16× 10−5), while sequevar 54 was significantly overrepresented in moderately elevated areas (R2) (χ2 test, P = 4.0× 10−3).
Pathogenicity of R. solanacearum isolates
According to different elevations, sequevars, and frequency, six representative strains were chosen for the pathogenicity study (Table 5). All tested strains were highly virulent on tobacco at tropical temperatures (30 °C day/25 °C night). Tobacco plants inoculated with R. solanacearum strains started to wilt 4 to 6 days after inoculation, and almost all inoculated plants were dead within 4 weeks (Fig. 4). Based on the final disease index, there was no difference in disease severity among the tested strains.

Severity of bacterial wilt on tobacco after inoculation with different strains of Ralstonia solanacearum isolated from China. Disease indices: 0 = asymptomatic plant, 1 = less than 25 % wilted leaves, 2 = less than 50 % wilted leaves, 3 = more than 50 % wilted leaves, and 4 = completely wilted leaves (dead plant). Error bars indicate standard errors of the means of two repeated experiments
Discussion
In this study, a total of 97 R. solanacearum isolates were isolated from four tobacco-growing zones over a wide range of elevations in China. All of these isolates produced typical R. solanacearum type colonies on TZC medium. We analyzed the genetic diversity of these R. solanacearum strains using phylotype and phylogenetic analysis. The results suggested that tobacco-infecting R. solanacearum strains all belonged to phylotype I and had sequevar diversity. The phylotype I strain was generally considered a tropical strain mostly prevalent in the lowlands; however, the finding of our study indicated that tobacco-infecting phylotype I strains were observed in China not only in the lowlands but also in moderately elevated areas and highlands. Phylotype I strains which infected potato were also reported from highland elevations in India (Sagar et al. 2014).
Phylotype I was known as the lineage with highest evolutionary potential because of its high ability of recombination, pattern of dissemination, large host range, and virulence plasticity. When retracing the complex evolutionary history of R. solanacearum using multilocus sequence analysis (MLSA), Wicker et al. (2012) reported that phylotype I was one of the ongoing diversifying subspecies. This study revealed that all tested tobacco phylotype I strains could be further clustered into eight egl-groups (sequevar), containing sequevars 13, 14, 15, 17, 34, 44, 54, and 55 (a new one) (Fig. 1). Among the eight sequevars, sequevar 13, 14, 15, 17, 34, 44, and 54 had been reported in Chinese tobacco R. solanacearum strains by Li et al. (2016), and sequevar 55, the new one, has not been previously described. The new sequevar may be an evidence that verified the ongoing evolutionary of phylotype I.
Southeast China (Z3), a warm and humid zone (Table 1), had the largest number of egl-groups and contained six sequevars: 13, 14, 15, 17, 34, and 44. In the Basin of the Yangzi River (Z1), there were five egl-groups: sequevars 15, 17, 34, 44, and 54. In southwest China (Z2), there were also five egl-groups containing sequevars 13, 17, 44, 54, and 55. In the basin of the Huai River (Z4), near the north of China, where slight bacterial wilt occurred recently, only sequevar 15 was identified, but whether the zone only has sequevar 15 requires identification of more isolates. The difference in sequevar distribution across tobacco-growing zones may be a result of the different conditions of temperature and rainfall (Table 1). A previous study reported that R. solanacearum was detected in river water and irrigation water and introduced the strains to cultivated fields in Europe and the United States (López and Biosca 2005; Hong et al. 2008). Paddy-upland rotation is an important measure in southeast China, especially in Fujian province. The other reason for the wide distribution and high diversity of R. solanacearum strains in southeast China may be attributed to cultivation measures and irrigation water. However, more evidence about the influence of paddy-upland rotation is required.
After analyzing the distribution of sequevars in different elevation areas, we knew that the genetic structure was associated with elevation. This study complemented previous reports on distinct types of R. solanacearum strains distributed at different elevations and agro-ecological zones (Perez et al. 2008; Mahbou Somo Toukam et al. 2009). It is noteworthy that sequevar 15 was clearly lowland-associated (22 of 25 sequevar 15 strains in R2) and predominant in southeast China. Taiwan, near southeast China, also had similary results that sequevar 15 was the most predominant one (Lin et al. 2014). The strains that were isolated from the highland elevations above 1500 m a.s.l. contained sequevar 17, 44, 54 and 55. It was surprising that the sequevar 54 (reported on tobacco R. solanacearum strains recently) (Li et al. 2016) and the new sequevar 55 were only found in areas with moderate to high elevations. The new sequevar may gather R.solanacearum strains evolving to adapt to the high elevations environment.
A pathogenicity test of the isolates on tobacco showed that all tested strains were strongly pathogenic at tropical lowland temperatures (30 °C day/25 °C night). Cellier and Prior (2010) reported that phylotype I strains were not virulent at cold temperate (15 and 24 °C, night and day, respectively), but it now remains to be determined whether the Chinese phylotype I strains spread to cold areas. As a follow-up to this study, the virulence of the highland strains to different hosts at cold temperatures is being examined to investigate the phylotype I strains associated with cool temperatures.
In summary, our study provides evidence that phylotype I strains have sequevar diversity and that genetic structure is associated with elevation in China, which is a prerequisite for effective breeding and management programs. That would inspirit growers for integrative control strategies and scientists for the in-depth thinking of local genetic diversity of R. solanacearum.
References
Boucher, C. A., Barberis, P. A., & Demery, D. A. (1985). Transposon mutagenesis of Pseudomonas solanacearum: isolation of Tn5-induced avirulent mutants. Microbiology, 131(9), 2449–2457.
Buddenhagen, I. W., Sequeira, L., & Kelman, A. (1962). Designation of races in Pseudomonas solanacearum. Phytopathology, 52(8), 726.
Castillo, J. A., & Greenberg, J. T. (2007). Evolutionary dynamics of Ralstonia solanacearum. Applied and Environmental Microbiology, 73(4), 1225–1238.
Cellier, G., & Prior, P. (2010). Deciphering phenotypic diversity of Ralstonia solanacearum strains pathogenic to potato. Phytopathology, 100(11), 1250–1261.
Cook, D., Barlow, E., & Sequeira, L. (1989). Genetic diversity of Pseudomonas solanacearum: detection of restriction fragment length polymorphisms with DNA probes that specify virulence and the hypersensitive response. Molecular Plant-Microbe Interactions, 2, 113–121.
Deberdt, P., Guyot, J., Coranson-Beaudu, R., Launay, J., Noreskal, M., Riviere, P., et al. (2014). Diversity of Ralstonia solanacearum in French Guiana expands knowledge of the "emerging ecotype. Phytopathology, 104(6), 586–596. doi:10.1094/Phyto-09-13-0264-R.
Fegan, M., & Prior, P. (2005). How complex is the "Ralstonia solanacearum species complex"? In C. Allen, P. Prior, & A. C. Hayward (Eds.), Bacterial wilt disease and the Ralstonia solanacearum species complex (pp. 449–462). St. Paul: APS.
Fegan, M., & Prior, P. (2006). Diverse members of the Ralstonia solanacearum species complex cause bacterial wilts of banana. Australasian Plant Pathology, 35(2), 93–101.
Frey, P., Smith, J. J., Albar, L., Prior, P., Saddler, G. S., Trigalet-Demery, D., et al. (1996). Bacteriocin typing of Burkholderia (Pseudomonas) solanacearum race 1 of the French west Indies and correlation with genomic variation of the pathogen. Applied and Environmental Microbiology, 62(2), 473–479.
Genin, S., & Denny, T. P. (2012). Pathogenomics of the Ralstonia solanacearum species complex. Annual Review of Phytopathology, 50, 67–89.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Hayward, A. C. (1964). Characteristics of Pseudomonas solanacearum. Journal of Applied Bacteriology, 27(2), 265–277.
Hayward, A. C. (1991). Biology and epidemiology of bacterial wilt caused by Pseudomonas solanacearum. Annual Review of Phytopathology, 29, 65–87.
He, L. Y., Sequeira, L., & Kelman, A. (1983). Characteristics of strains of Pseudomonas solanacearum from China. Plant Disease, 67(12), 1357–1361.
Hong, J. C., Momol, M. T., Jones, J. B., Ji, P. S., Olson, S. M., Allen, C., et al. (2008). Detection of Ralstonia solanacearum in irrigation ponds and aquatic weeds associated with the ponds in North Florida. Plant Disease, 92(12), 1674–1682.
Horita, M., & Tsuchiya, K. (2001). Genetic diversity of Japanese strains of Ralstonia solanacearum. Phytopathology, 91(4), 399–407.
Jeanmougin, F., Thompson, J. D., Gouy, M., Higgins, D. G., & Gibson, T. J. (1998). Multiple sequence alignment with Clustal X. Trends in Biochemical Sciences, 23(10), 403–405.
Kelman, A. (1954). The relationship of pathogenicity in Pseudomonas Solanacearum to colony appearance on a tetrazolium chloride medium. Phytopathology, 44(12), 693–695.
Kong, F. (2003). Integrated control of tobacco bacterial wilt disease. Tobacco science and Technology, 4, 42–43 (In Chinese).
Lebeau, A., Gouy, M., Daunay, M. C., Wicker, E., Chiroleu, F., Prior, P., et al. (2013). Genetic mapping of a major dominant gene for resistance to Ralstonia solanacearum in eggplant. Theoretical and Applied Genetics, 126(1), 143–158. doi:10.1007/s00122–012–1969-5.
Li, X., Dorsch, M., Del Dot, T., Sly, L., Stackebrandt, E., & Hayward, A. (1993). Phylogenetic studies of the rRNA group II pseudomonads based on 16S rRNA gene sequences. Journal of Applied Bacteriology, 74(3), 324–329.
Li, Y., Feng, J., Liu, H., Wang, L., Hsiang, T., Li, X., & Huang, J. (2016). Genetic Diversity and Pathogenicity of Ralstonia solanacearum Causing Tobacco Bacterial Wilt in China. Plant Disease, 100(7), 1288–1296.
Lin, C. H., Tsai, K. C., Prior, P., & Wang, J. F. (2014). Phylogenetic relationships and population structure of Ralstonia solanacearum isolated from diverse origins in Taiwan. Plant Pathology, 63(6), 1395–1403. doi:10.1111/Ppa.12209.
López, M., & Biosca, E. (2005). Potato bacterial wilt management new prospects for an old problem. In C. Allen, P. Prior, & A. C. Hayward (Eds.), Bacterial wilt disease and the Ralstonia solanacearum species complex (pp. 205–224). St. Paul: APS.
Lu, T. (1998). Advances in research of pseudomonas solanacearum of crop in China. Fujian The Journal of Agricultural Science, 13(2), 33–40. (In Chinese)
Mahbou Somo Toukam, G., Cellier, G., Wicker, E., Guilbaud, C., Kahane, R., Allen, C., et al. (2009). Broad diversity of Ralstonia solanacearum strains in Cameroon. Plant Disease, 93(11), 1123–1130. doi:10.1094/pdis-93-11-1123.
N’guessan, C., Abo, K., Fondio, L., Chiroleu, F., Lebeau, A., Poussier, S., et al. (2012). So near and yet so far: the specific case of Ralstonia solanacearum populations from Cote d’Ivoire in Africa. Phytopathology, 102(8), 733–740.
Opina, N., Tavner, F., Hollway, G., Wang, J. F., Li, T. H., Maghirang, R., et al. (1997). A novel method for development of species and strain-specific DNA probes and PCR primers for identifying Burkholderia solanacearum (formerly Pseudomonas solanacearum). Asia-Pacific Journal of Molecular Biology and. Biotechnology, 5(1), 19–30.
Perez, A. S., Mejia, L., Fegan, M., & Allen, C. (2008). Diversity and distribution of Ralstonia solanacearum strains in Guatemala and rare occurrence of tomato fruit infection. Plant Pathology, 57(2), 320–331. doi:10.1111/j.1365–3059.2007.01769.x.
Poussier, S., Prior, P., Luisetti, J., Hayward, A. C., & Fegan, M. (2000a). Partial sequencing of the hrpB and Endoglucanase genes confirms and expands the known diversity within the Ralstonia solanacearum species complex. Systematic and Applied Microbiology, 23(4), 479–486. doi:10.1016/s0723–2020(00)80021–1.
Poussier, S., Trigalet-Demery, D., Vandewalle, P., Goffinet, B., Luisetti, J., & Trigalet, A. (2000b). Genetic diversity of Ralstonia solanacearum as assessed by PCR-RFLP of the hrp gene region, AFLP and 16S rRNA sequence analysis, and identification of an African subdivision. Microbiology, 146, 1679–1692.
Prior, P., & Fegan, M. (2005). Recent developments in the phylogeny and classification of Ralstonia solanacearum. Acta Horticulturae, 695, 127–136.
Ramesh, R., Achari, G. A., & Gaitonde, S. (2014). Genetic diversity of Ralstonia solanacearum infecting solanaceous vegetables from India reveals the existence of unknown or newer sequevars of Phylotype I strains. European Journal of Plant Pathology, 140(3), 543–562. doi:10.1007/s10658–014–0487-5.
Sagar, V., Jeevalatha, A., Mian, S., Chakrabarti, S. K., Gurjar, M. S., Arora, R. K., et al. (2014). Potato bacterial wilt in India caused by strains of phylotype I, II and IV of Ralstonia solanacearum. European Journal of Plant Pathology, 138(1), 51–65.
Siri, M. I., Sanabria, A., & Pianzzola, M. J. (2011). Genetic diversity and aggressiveness of Ralstonia solanacearum strains causing bacterial wilt of potato in Uruguay. Plant Disease, 95(10), 1292–1301.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., & Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28(10), 2731–2739.
Wicker, E., Grassart, L., Coranson-Beaudu, R., Mian, D., Guilbaud, C., Fegan, M., et al. (2007). Ralstonia solanacearum strains from Martinique (French west indies) exhibiting a new pathogenic potential. Applied and Environmental Microbiology, 73(21), 6790–6801.
Wicker, E., Lefeuvre, P., de Cambiaire, J. C., Lemaire, C., Poussier, S., & Prior, P. (2012). Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME Journal, 6(5), 961–974.
Xu, J., Pan, Z. C., Prior, P., Xu, J. S., Zhang, Z., Zhang, H., et al. (2009). Genetic diversity of Ralstonia solanacearum strains from China. European Journal of Plant Pathology, 125(4), 641–653.
Xue, Q. Y., Yin, Y. N., Yang, W., Heuer, H., Prior, P., Guo, J. H., & Smalla, K. (2011). Genetic diversity of Ralstonia solanacearum strains from China assessed by PCR-based fingerprints to unravel host plant-and site-dependent distribution patterns. FEMS Microbiology Ecology, 75(3), 507–519.
Zhou, X., Wang, J., Yang, Y., Zhao, T., & Gao, B. (2012). Advances in tobacco bacterial wilt disease. Microbiology China, 39(10), 1479–1486 (In Chinese).
Zhu, C., Wang, Y., & Wang, Z. (2002). Tobacco Diseases of China. Beijing: China Agriculture Press (In Chinese).
Acknowledgments
This research was supported by the Key Project from China National Tobacco Corporation (110201202002) and the Key Project from China National Tobacco Corporation Chongqing Branch (NY20130501070005).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Liu, Y., Wu, D., Liu, Q. et al. The sequevar distribution of Ralstonia solanacearum in tobacco-growing zones of China is structured by elevation. Eur J Plant Pathol 147, 541–551 (2017). https://doi.org/10.1007/s10658-016-1023-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-016-1023-6