
Abstract
The Pleistocene ice age and recent forest fragmentation have both played a significant role in shaping the population genetic variation of boreal coniferous species in the Qinghai-Tibet Plateau (QTP) and Eurasian coniferous forests. The Sichuan jay is one of the least know endemic bird species in QTP coniferous forests in western China while its sibling species, the Siberian Jay, is widespread within the coniferous forests in northern Eurasia. Here we used 11 microsatellite markers to assay genetic diversity across 58 Sichuan jay samples from China and 205 Siberian jay samples from Sweden and Russia. Results showed three distinct genetic clusters from the Sichuan jay sampling. Furthermore, the pair-wise FST values indicated high genetic differentiation not only among the two species but also between Swedish and Russian Siberian jay populations. What is more, a pattern of isolation by distance was found among the analyzed populations. Our study suggests that targeted habitat restoration in fragmented forests and more genetic work is urgently needed for conservation of the Sichuan jay.
Similar content being viewed by others

Introduction
Forest fragmentation has profound and lasting influences around the globe, not only on the Earth’s biological diversity, but on ecosystem function and numerous ecosystem services (Kettle and Koh 2014; Pfeifer et al. 2017). At present, more and more large areas of continuous forests of the world are rapidly becoming fragmented by human influence, which apparently has negative ecological and genetic effects in the resulting forest patches for species that dwell in isolated forest islands (Haddad et al. 2015; Nason and Hamrick 1997; Robinson et al. 1995). Moreover, the direct ecological consequences of forest fragmentation include changing habitat connectivity, reducing species’ richness, availability, and specialization (Bregman et al. 2014; Hadley et al. 2018; Hernando et al. 2017).
Fragmentation of continuous forests to become isolated patches can pose a long-term threat to populations and species through genetic bottlenecks, ecological traps, changing patterns of selection, inbreeding, drift, and gene flow, especially in the smallest and most isolated fragments (Bacles et al. 2005; Gaines et al. 1997; Höglund 2009; Orrock 2005; Templeton et al. 1990; Young et al. 1996). In addition to anthropogenic influence, mountain uplifts and past climatic changes have also caused forest fragmentation, which have resulted in isolation, divergence, and speciation in face of reduced gene flow in populations and species inhabiting such areas (Filatov et al. 2016; Rice and Hostert 1993). For example, the expanding ice sheets directly fragmented the boreal forest during recent glacial periods (Weir and Schluter 2004).
Speciation and divergence in boreal species promoted by natural causes are common in the coniferous forests of the Qinghai-Tibet Plateau (QTP), Northern Eurasia and North America (Hewitt 1996; Baba et al. 2002; Weir and Schluter 2004; Zhan et al. 2011). Although there is controversy about the role of the Quaternary ice ages in vertebrate speciation across the globe, the geographical distributions of some boreal species have indeed been fragmented periodically by habitat changes accompanying global cooling. This is related to the extent of boreal forest, which is a geographically extensive habitat zone and which was directly fragmented by advancing glaciers into multiple refugia (Avise 1998; Hewitt 2000, 2004; Zink and Slowinski 1995). There are several studies of genetic patterns in the northern boreal forest avifaunas in Europe and North America (Corrales et al. 2014; Höglund et al. 2013; Weir and Schluter 2004; Zhan et al. 2011). However, few studies have been conducted on sibling species in QTP and eastern and western Siberia, especially for those species that have poor dispersal capability (Lu et al. 2012a; Sun et al. 2001). Studies of this isolated avifauna should thus yield a greater understanding of the speciation into sibling species following fragmentation. During Pleistocene, many avian species living in boreal forest have evolved into superspecies or to different subspecies after they experienced allopatric divergence and local adaptation (Davis and Shaw 2001; Hewitt 2004; Li et al. 2013; Ren et al. 2017). High altitude boreal forests were preserved in the southeast edge of QTP by the uplift of QTP which comprise one of the key high-altitude biodiversity hotspots in the world (Myers et al. 2000). The evolutionary history and genetic diversity of the sibling species in QTP and Siberian boreal forests have not yet been fully examined.
In addition to ice ages affecting boreal fragmentation, more recent human actions, for instance, heavy deforestation, have exacerbated the fragmentation of boreal forest (Wallenius et al. 2010). The high-elevation coniferous forest, an important and special part of boreal forests, are located at the eastern and southern rim of QTP. This forest acts as a modern harbour for many rare species because of its relatively pristine state (Cincotta et al. 2000; Liu et al. 2009), such as Chinese grouse (Tetrastes sewerzowi) (Sun 2000), Blood pheasant (Ithaginis cruentus) (Jia et al. 2010) and Wild yak (Bos grunniens) (Ceballos et al. 1993). However, local species in this montane ecosystems are sensitive to the effects of human activity and climate change (Liu et al. 2009; Lyu and Sun 2014; Pounds et al. 1999).
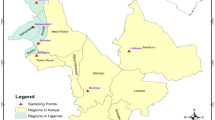
The Sichuan Jay (Perisoreus internigrans) is one of the least known endemic bird species in western China and inhabits high mountain coniferous forests at an altitude between 2800 and 4500 m. It is a sibling species to the Siberian jay (P. infaustus) which inhabits boreal forest from Eastern Siberia to Fenno-Scandia in the west. The Sichuan jay has been recorded from only 17 locations in Gansu, Sichuan, Qinghai and Tibet (Fig. 1) (Jing 2005; Sun et al. 2001). The species is qualified as “Vulnerable” in IUCN’s red list as it has a small, declining, severely fragmented populations as a result of extensive deforestation throughout its range (BirdLife International 2020). Despite the small and declining populations, it has not been put on the national protection list in China.

Current distribution of Sichuan jay (Perisoreus internigrans) in southwest China (light gray), and Siberian jay (Perisoreus infaustus, dark gray), and the locations of sampling sites (black dots)
The first reliable survey of numbers and distribution was conducted from 1999–2004 in the forest areas in Zhuoni, Gansu (34° 14′–34° 38′ N 103° 12′–103° 32′ E) and Jiuzhaigou, Sichuan (33° 31′–33° 7′ N 103° 39′–103° 46′ E) (Yu et al. 2011). The two locations are about 120 km apart, and the forest areas are 20,000 and 16,000 ha, respectively. At the Zhuoni site, most of the old-growth spruce (Picea spp.) and fir (Abies spp.) were removed in selective cuts by the government in the 1930s and 1990s. At the Jiuzhaigou site, patches of pristine of forest alternate with clear-cut replanted areas (Sun et al. 2001). In these areas only a few small Sichuan jay populations were found, although the exact numbers of individuals were not obtained. Population density in Jiuzhaigou was about 1.04 per km2, and was even lower in Zhuoni (Yu et al. 2011). Until now, suitable Sichuan Jay habitat became increasingly fragmented from 1950s and very little was known regarding the genetics of this globally threatened montane bird (Jing et al. 2009; Lu et al. 2012b).
Here we use microsatellite allele frequencies to report: (1) genetic variation among endangered Sichuan jay populations; (2) compare levels of genetic diversity in the Sichuan jay population with Siberian jay populations from Siberia and Sweden; (3) estimate the effective population size and isolation by distance (IBD) of the Sichuan jay and Siberian jay; (4) a better understanding of the history and background data of this two sibling species, which could be used as a guideline for conservation of the Sichuan jay and Siberian jay and for other sibling species with similar characteristics.
Methods
DNA samples
In total, 58 Sichuan jay samples were obtained during 1999 to 2003 from China and 205 Siberian jay samples were collected during 2008 to 2010 from Sweden and Russia. For Sichuan jay samples, blood samples (N = 30) and feather or muscle samples (N = 28) from 58 captured jays and predation remains were collected in Sichuan and Gansu, western China (Fig. 1). Blood and muscle samples were stored in absolute ethanol in the field, and then stored long term at − 20℃ in the lab. Feathers were stored dry at room temperature. DNA was extracted using a DNeasy® Tissue kit (QIAGEN, https://www.qiagen.com/) following the manufacturer’s procedures. Siberian jay blood samples came from three populations in Russia Siberia (i.e. Burenski; Kolyma and Yakutsk) and from two populations in Sweden (i.e. Ånnsjön and Vilhelmina). DNA was extracted using a standard high salt extraction protocol modified from Paxton et al. (1996).
Molecular methods
Twenty one microsatellite loci originally developed for Siberian jay were amplified (Jaari et al. 2008) in both our Sichuan jay and Siberian jay samples. The microsatellite markers were divided into groups according to fluorescent dyes, which were FAM (SJ013, SJ014, SJ016, SJ026, SJ046, SJ049, SJ059, SJ068, SJ070, SJ078, SJ081, SJ083, SJ092, SJ093, SJ095, SJ106, SJ109, SJ116) and NED (SJ058, SJ065, SJ099). For the Sichuan jays, PCRs were run in 10 µL reactions containing 0.20 µL of each primer (10 mM), 1.00 µL of DNA, 1.00 µL 10 × PCR buffer, 0.50 µL dNTPs (2.5 mM), 0.30 µL BSA, 0.45 µL MgCl2 (25 mM), 0.05 µL DreamTaq and 6.50 µL water. PCR programs were an initial denaturation cycle at 95 °C for 3 min, followed by 5 cycles at 95 °C for 30 s, annealing at 59 °C to 54 °C for 30 s and 72 °C for 1 min, then followed by 30 cycles at 95 °C for 30 s, annealing at 54 °C for 30 s and 72 °C for 30 s. The PCRs had a final extension at 72 °C for 10 min. The PCR products were diluted in water (1.1:10). 1 μl of the dilution was mixed with 10 μl formamid/H2O containing 0.1 µl GeneScanTM 600 LIZ® size standard (Thermo Scientific) and run on 3730xl DNA Analyzer (Applied Biosystems). Samples were genotyped using GeneMapper® Software 5 (Life Technologies™). For the Siberian Jays, we amplified microsatellites makers using Qiagen multiplex mix according to the manufacturers’ recommendations. Amplified products were diluted 1:100 with Megabace ET400-R as size standard prior to analysis on a Megabace 1000 (GE healthcare/Amersham) and genotyping was conducted using Megabace Fragment profiler 1.2 (GE healthcare/Amersham).
In total, 21 microsatellite loci amplified a PCR product giving genotypes for the 58 Sichuan jay samples. As SJ046 was found to be linked to the Z-chromosome, it was removed from further analyses. We used the Excel add-in Microsatellite toolkit to convert input microsatellite data files for different analyses program formats (Park 2001). All the microsatellite markers were analyzed with MICROCHECKER 2.2.3 (Van Oosterhout et al. 2004) to check for null alleles. Six out of 20 microsatellite loci (SJ013, SJ026, SJ065, SJ081, SJ083, and SJ092) were found to contain null alleles and were thus removed from further analyses. SJ014 and SJ059 were not available for Siberian jays. We tested for Hardy and Weinberg equilibrium and linkage disequilibria between molecular markers. To ensure the neutrality and independently segregating markers we discarded SJ016, SJ049 and SJ116. Thus, we used 11 microsatellite loci to analyze Sichuan jay samples. However, we used only 9 of the same microsatellite markers out of the previous 11 to analyze the Siberian jay samples due to lack of amplification concerning the makers SJ014 and SJ059.
Statistical analysis
We calculated expected heterozygosity (He), observed heterozygosity (Ho), FIS, allelic richness (AR), and effective population size (Ne) in each of the sampled populations. The neutral genetic diversity for each locality was calculated in FSTAT 2.9.3.2 (Goudet 1995). Effective population size was estimated based on two different methods: the linkage disequilibrium method and the coancestry method using the software Ne estimator (Waples and Do 2008).
To illustrate the multidimensional relationships among individual Sichuan jays in a two-dimensional plot, we used the R package ADEGENET (Jombart 2008) to create discriminant analysis of principal components (DAPC) with 100,000 iterations and 10,000 burn-in steps. In addition we performed Bayesian clustering using STRUCTURE v.2.3.4 (Pritchard et al. 2000) to find genetic clusters among the all samples and for Sichuan jays excluding Siberian jays. The dataset was run ten times per K = 1–10 with an initial burn-in of 10,000 followed by 100,000 MCMC iterations and 100,000 burn-in followed by 100,000 MCMC iterations for Sichuan jays only. Visualization of the runs was done using R package POPHELPER v2.2.9 (Francis 2017) after averaging over all STRUCTURE runs for each K using CLUMPP (Jakobsson and Rosenberg 2007) included in the POPHELPER package.
To calculate population differentiation between the sampled locations, we used the data from the 9 microsatellite loci common to both species to calculate pair-wise FST (Weir and Cockerham 1984) and Nei’s genetic distance using SpaGeDi v1.5 (Hardy and Vekemans 2002). With their geographic coordinates, we calculated the Euclidean distance as the geographical distances. Then we used the geographical distance and FST/(1 − FST) to calculate the IBD in SpaGeDi v 1.5 (Hardy and Vekemans 2002).
Tests of past population bottlenecks were performed by calculating the difference between the expected heterozygosity under Hardy–Weinberg equilibrium (HE) and the heterozygosity expected at mutation-drift equilibrium (Heq) assuming three different mutation models, the stepwise mutation model (SMM), two-phased models (TPM) and the infinite allele model (IAM), as implemented in software Bottleneck (Piry et al. 1999). We used Wilcoxon sign rank test to obtain P values. The TPM was tested in different conditions with respect to the proportion of step-wise mutations (70, 60, 50, 40, 30, 20 and 10%).
Results
Genetic variation
In the entire Sichuan jay population, expected heterozygosity (He) was 0.741, observed heterozygosity (Ho) was 0.815, and allelic richness (AR) was 3.25 (Table 1). We calculated a reliable value for Ne by the linkage disequilibrium method (Ne (LDNe)) and by the coancestry method (CNe). The estimates were: 16.1 (95% CI 9.0 to 25.2) and 39.9 (95% CI 31.9 to 51.3), respectively (Table 1).
In Siberian jays, He within the different populations ranged from 0.675 to 0.801, Ho from 0.627–0.750 and allelic richness from 2.91 to 3.48. Observed and expected heterozygosity were not significantly different, as would be expected of selectively neutral loci. Thus, based on the nine common microsatellites makers, we found heterozygosity and allelic richness to be of the same magnitude in both species. Our estimates of effective population size revealed similar values among the sampled populations with the exception of the Siberian jays in Kolyma which showed a significantly higher estimate (Table 1).
Population differentiation
Geographic structure and separation among the sampled Sichuan jay was supported by the discriminant analysis of principle components (DAPC) (Fig. 2) which shows three main clusters among the Chinese samples based on the Bayesian Information Criterion. These results were confirmed by STRUCTURE analyses which support three weakly differentiated clusters when sorted by assignment probability (Q) (Suppl. Fig. 1). For all jay samples, three distinct clusters was found in the STRUCTURE analysis (Suppl. Fig. 2), supported by the Evanno method (Evanno et al. 2005) that gave the highest support for K = 3 (delta − K) (Suppl. Fig. 3). The three clusters followed country of sampling.

Discriminant analysis of principle components (DAPC) scatterplot. The three DAPC clusters are based on 11 microsatellite makers from 58 P. infaustus from Sichuan, China
Pair-wise FST values ranged between 0.026 and 0.52 when all populations and both species were included in the analysis (Table 2). As expected, the highest FST values were found between Sichuan jays and Siberian jay populations as they belong to different species. With the exception of the Kolyma–Sichuan comparison the overall FST were not much higher than the comparison between Swedish and Russian Siberian jay populations. In contrast, the within region comparisons in Sweden and Russia had the lowest values and comparisons between these regions had the highest FST values respectively. Nei’s genetic distance values had the same trend as the FST values (Table 2). The lowest values were found between the two sampled locations from Sweden and three sampled locations from Russia. The highest values were found between Sichuan jays and Siberian jays (Table 2).
A highly significant correlation was found between genetic differentiation and geographic distance of populations when all populations were included in the IBD analyses (r2 = 0.5113, P < 0.001; Fig. 3). When IBD analysis was run only on Siberian jay populations, the correlation was also highly significant (r2 = 0.8568, P < 0.001; Fig. 3).

Isolation by distance plots (IBD) for microsatellite data. FST/(1 − FST) is a function of distance (km). Black squares: IBD for pair-wise population comparisons of 263 individuals of P. internigrans and P. infaustus. Black circles: IBD based on 205 individuals of P. infaustus. The r2 represents the coefficient of determination and P the probability value. The dashed line: the trend curve for all jays; The dots line: the trend curve for Siberian jays
Analyses of population bottlenecks
We found evidence of mutation-drift heterozygote excess (Heq) according to a one-tailed Wilcoxson test for heterozygote excess in all populations, except when assuming a strict SMM (Table 3). We therefore conclude that there is a strong indication of past population bottlenecks in both species.
Discussion
The results of this study show that the Sichuan jay, which inhabit a narrow band of fragmented high-elevation coniferous forests, has similar levels of genetic variability as compared to the Siberian jay since estimates of genetic diversity were similar when comparing values of AR, Ho and He. These sister species formed after the rise of QTP during the Quaternary Period (Cheng 1981) as some other sibling species pairs which like jays have a similar distribution pattern, such as the Chinese grouse and hazel grouse (T. bonasia). That estimates of genetic diversity in Sichuan and Siberian jay populations are similar is somewhat surprising as we would have expected lower diversity in the more range restricted Sichuan jay.
The distribution and genetic diversity of species are important for their conservation, especially for relict populations in isolated refugia (Höglund 2009). As the first study describing the genetic diversity of Sichuan jay, the discriminant analysis revealed genetic differentiation among Sichuan jay samples (Fig. 2). Unfortunately the x–y coordinates of the Sichuan jay samples have been lost since the date of capture and we can thus not test if the detected substructure is due to geographically separated populations. However, as a proxy of gene flow rupture, the observed genetic differentiation confirm previous results showing that remaining Sichuan jay habitats are at present isolated and fragmented because of intensive forest cutting of the virgin forest during the past decades (Lu et al. 2012b; Sun 2000).
Ne is one of the most important parameters in conservation genetics and could be regarded as an estimate of genetic diversity since it corresponds to the heterozygosity in an ideal randomly mating population with the estimated size. A low Ne value is also associated with accelerated depletion of genetic variability (Hájková et al. 2007). In all these six populations, including both sub species, our estimates of Ne from two methods provided reliable values except for Kolyma which had a value reaching infinity using the CNe method, which is indicative of a too small sample size. The Ne/N ratio in wild populations is approximately estimated to be in the range of 0.10 (Frankham 1995). This estimate would for Sichuan jay in Sichuan give a population size of 162 and 399 individuals which is close to the range of 132 to 374 given by our census data from 2000 to 2004 (Jing 2005).
The pairwise FST and Nei’s genetic distance (Ds) analyses showed the highest FST values and Ds values between Sichuan and Siberian jay populations, and also showed high values when comparing populations from Sweden with Russia. Nei’s genetic distance analysis showed a strong divergence among the two species. The Sichuan jay population was separated from the five Siberian populations. Correspondingly there were high FST values among Swedish and Russian populations. Such genetic differentiation between QTP coniferous forest species and sister taxa residing in coniferous forest in northern Eurasia are common in other animals and insects confined to boreal forest (Baba et al. 2002; Johansson et al. 2017). For a conservation perspective, preserving boreal forest surrounding the QTP would also benefit the QTP-residing species of avina genera such as: Tetrastes, Strix, Aegolius and Picoides.
Our results indicate that IBD pattern was found not only in the total locations of Sichuan jay and Siberian jay, but also among Swedish and Russian locations of the Siberian jay (Fig. 3). This IBD pattern may be influenced by the fragmentation of boreal forest which was affected by the last ice age and recent human actions (Wallenius et al. 2010). The described situation should be similar in other boreal birds in Siberia and the QTP. Some studies have suggested that many of these populations, like hazel grouse, might come from one or a few refugial lineages from central and western coniferous forest in Eurasia, which might have been almost completely eradicated during the major glaciations (Baba et al. 2002; Englbrecht et al. 2000). The divergence and speciation in the boreal avifauna during this period resulted in some becoming sister-species, like the two jays studied here and some becoming subspecies like in boreal owl Aegolius funereus (Weir and Schluter 2004). Their dispersal abilities and response to the ice age and subsequent warming correspond to their divergence and speciation (Barluenga and Meyer 2005; Hewitt 1999). Large-scale comparative population genetic studies of sister-species, like Sichuan jay and Siberian jay, of boreal coniferous forest would provide a comprehensive view of the genetic consequences of the glaciation events on species with different life histories, and their effect on the current genetic diversity and geographical distribution patterns. Due to the glaciation events in the ancient and recent human actions, boreal forests became fragmented and isolated (Wallenius et al. 2010). Thus, we hypothesize that the jay populations of Sichuan and Siberia have experienced bottleneck as a result of habitat loss that likely occurred during glacial period and/or more recently as a result human disturbances.
Habitat loss is the most important cause of species extinction (IUCN 2020). In order to maintain healthy populations of Sichuan jay in western China, targeted habitat restoration is important which can reduce extinction rates in fragmented forests (Newmark et al. 2017). To prevent nest predation, that is the most common cause of nest failure in Siberian jays, they need to nest in virgin conifer forest for sucessful breeding (Eggers et al. 2005; Griesser and Nystrand 2009). As both species are expected to have similar breeding ecology and need prestine forest for sucessful breeding, it is of utter importance for the recrutiment of new individuals to the population, we therefore suggest that more attention should be paid to the protection of virgin conifer forest in western China. This will benefit not only the Sichuan jay, but also many other rare species distributed in the same area, such as Chinese grouse, Boreal owl, Sichuan Wood Owl, and blood pheasant. There is also an urgent need to study other populations of Sichuan jay in China and central Russian populations of Siberian jay not included in this study, to better understand the conservation value, needs and importance of each population.
References
Avise JC (1998) Pleistocene phylogeographic effects on avian populations and the speciation process. Proc R Soc Lond B 265:457–463
Baba Y, Fujimaki Y, Klaus S, Butorina O, Drovetskii S, Koike H (2002) Molecular population phylogeny of the hazel grouse Bonasa bonasia in East Asia inferred from mitochondrial control-region sequences. Wildl Biol 8:251–260
Bacles CF, Burczyk J, Lowe AJ, Ennos RA (2005) Historical and contemporary mating patterns in remnant populations of the forest tree Fraxinus excelsior L. Evolution 59:979–990
Barluenga M, Meyer A (2005) Old fish in a young lake: stone loach (Pisces: Barbatula barbatula) populations in Lake Constance are genetically isolated by distance. Mol Ecol 14:1229–1239
BirdLife International (2020) Species factsheet: Perisoreus internigrans. https://www.birdlife.org/ Accessed on 7 Feb 2020
Bregman TP, Sekercioglu CH, Tobias JA (2014) Global patterns and predictors of bird species responses to forest fragmentation: implications for ecosystem function and conservation. Biol Conserv 169:372–383
Ceballos G, Mellink E, Hanebury LR (1993) Distribution and conservation status of prairie dogs Cynomys mexicanus and Cynomys ludovicianus in Mexico. Biol Conserv 63:105–112
Cheng T-S (1981) The fauna and its evolution of territorial vertebrates at Qinghai-Tibet plateau. Rep Beijing Natl Mus 9:1–17
Cincotta RP, Wisnewski J, Engelman R (2000) Human population in the biodiversity hotspots. Nature 404:990
Corrales C, Pavlovska M, Höglund J (2014) Phylogeography and subspecies status of Black Grouse. J Ornithol 155:13–25
Davis MB, Shaw RG (2001) Range shifts and adaptive responses to quaternary climate change. Science 292:673–679
Eggers S, Griesser M, Andersson T, Ekman J (2005) Nest predation and habitat change interact to influence Siberian jay numbers. Oikos 111(1):150–158
Englbrecht C, Freyhof J, Nolte A, Rassmann K, Schliewen U, Tautz D (2000) Phylogeography of the bullhead Cottus gobio (Pisces: Teleostei: Cottidae) suggests a pre-Pleistocene origin of the major central European populations. Mol Ecol 9:709–722
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Filatov DA, Osborne OG, Papadopulos AS (2016) Demographic history of speciation in a Senecio altitudinal hybrid zone on Mt. Etna. Mol Ecol 25:2467–2481
Francis RM (2017) Pophelper: an R package and web app to analyse and visualize population structure. Mol Ecol Resour 17:27–32. https://doi.org/10.1111/1755-0998.12509
Frankham R (1995) Conservation genetics. Annu Rev Genet 29:305–327
Gaines M, Diffendorfer J, Tamarin R, Whittam T (1997) The effects of habitat fragmentation on the genetic structure of small mammal populations. J Hered 88:294–304
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Griesser M, Nystrand M (2009) Vigilance and predation of a forest-living bird species depend on large-scale habitat structure. Behav Ecol 20(4):709–715
Haddad NM et al (2015) Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci Adv 1:e1500052
Hadley AS, Frey SJ, Robinson WD, Betts MG (2018) Forest fragmentation and loss reduce richness, availability, and specialization in tropical hummingbird communities. Biotropica 50:74–83
Hájková P, Pertoldi C, Zemanová B, Roche K, Hajek B, Bryja J, Zima J (2007) Genetic structure and evidence for recent population decline in Eurasian otter populations in the Czech and Slovak Republics: implications for conservation. J Zool 272(1):1–9
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Resour 2:618–620
Hernando A, Velázquez J, Valbuena R, Legrand M, García-Abril A (2017) Influence of the resolution of forest cover maps in evaluating fragmentation and connectivity to assess habitat conservation status. Ecol Ind 79:295–302
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Lin Soc 58(3):247–276
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Lin Soc 68:87–112
Hewitt G (2000) The genetic legacy of the quaternary ice ages. Nature 405:907
Hewitt G (2004) Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc B 359:183–195
Höglund J (2009) Evolutionary conservation genetics. Oxford University Press, New York
Höglund J, Wang B, Axelsson T, Quintela M (2013) Phylogeography of willow grouse (Lagopus lagopus) in the Arctic: taxonomic discordance as inferred from molecular data. Biol J Lin Soc 110:77–90
IUCN (2020) The IUCN red list of threatened species. Version 2019-3. https://www.iucnredlist.org.
Jaari S, Välimäki K, Merilä J (2008) Isolation and characterization of 100 polymorphic microsatellite loci for the Siberian jay (Perisoreus infaustus). Mol Ecol Resour 8:1469–1474
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jia C-X, Sun Y-H, Swenson JE (2010) Unusual incubation behavior and embryonic tolerance of hypothermia by the Blood Pheasant (Ithaginis cruentus). Auk 127:926–931
Jing Y (2005) Life history, cooperative breeding, foraging competition and distribution of Sichuan jay (Perisoreus internigrans). Institute of Zoology, Chinese Academy of Sciences, Beijing
Jing Y, Fang Y, Strickland D, Lu N, Sun Y-H (2009) Alloparenting in the rare Sichuan jay (Perisoreus internigrans). Condor 111:662–667
Johansson F, Halvarsson P, Mikolajewski D, Höglund J (2017) Genetic differentiation in the boreal dragonfly Leucorrhinia dubia in the Palearctic region. Biol J Lin Soc 121:294–304
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Kettle CJ, Koh LP (2014) Global forest fragmentation. CABI, Wallingford. https://doi.org/10.1079/9781780642031.0050
Li L et al (2013) Pliocene intraspecific divergence and Plio-Pleistocene range expansions within Picea likiangensis (Lijiang spruce), a dominant forest tree of the Qinghai-Tibet Plateau. Mol Ecol 22:5237–5255
Liu Z et al (2009) The effect of landscape features on population genetic structure in Yunnan snub-nosed monkeys (Rhinopithecus bieti) implies an anthropogenic genetic discontinuity. Mol Ecol 18:3831–3846
Lu N, Jia C-X, Lloyd H, Sun Y-H (2012a) Species-specific habitat fragmentation assessment, considering the ecological niche requirements and dispersal capability. Biol Conserv 152:102–109
Lu N, Jing Y, Lloyd H, Sun Y-H (2012b) Assessing the distributions and potential risks from climate change for the Sichuan jay (Perisoreus internigrans). Condor 114:365–376
Lyu N, Sun Y-H (2014) Predicting threat of climate change to the Chinese grouse on the Qinghai–Tibet plateau. Wildl Biol 20:73–82
Myers N, Mittermeier RA, Mittermeier CG, Da Fonseca GA, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853
Nason J, Hamrick J (1997) Reproductive and genetic consequences of forest fragmentation: two case studies of neotropical canopy trees. J Hered 88:264–276
Newmark WD, Jenkins CN, Pimm SL, McNeally PB, Halley JM (2017) Targeted habitat restoration can reduce extinction rates in fragmented forests. Proc Natl Acad Sci 114:9635–9640
Orrock JL (2005) Conservation corridors affect the fixation of novel alleles. Conserv Genet 6:623–630
Park S (2001) The Excel microsatellite toolkit trypanotolerance in West African Cattle and the population genetic effects of selection
Paxton RJ, Thorén PA, Tengö J, Pamilo P (1996) Mating structure and nestmate relatedness in a communal bee, Andrena jacobi (Hymenoptera, Andrenidae), using microsatellites. Mol Ecol 5(4):511–519
Pfeifer M et al (2017) Creation of forest edges has a global impact on forest vertebrates. Nature 551:187
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Pounds JA, Fogden MP, Campbell JH (1999) Biological response to climate change on a tropical mountain. Nature 398:611
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Ren G et al (2017) Genetic consequences of quaternary climatic oscillations in the Himalayas: Primula tibetica as a case study based on restriction site-associated DNA sequencing. New Phytol 213:1500–1512
Rice WR, Hostert EE (1993) Laboratory experiments on speciation: what have we learned in 40 years? Evolution 47:1637–1653
Robinson SK, Thompson FR III, Donovan TM, Whitehead DR, Faaborg J (1995) Regional forest fragmentation and the nesting success of migratory birds. Science 267:1987
Sun Y-H (2000) Distribution and status of the Chinese grouse Bonasa sewerzowi. Wildl Biol 6:271–275
Sun YH, Jia CX, Fang Y (2001) The distribution and status of Sichuan Grey Jay (Perisoreus internigrans). J Ornithol 142:93–98
Templeton AR, Shaw K, Routman E, Davis SK (1990) The genetic consequences of habitat fragmentation. Ann Mo Bot Gard 77:13–27
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Resour 4:535–538
Wallenius T et al (2010) Loss of habitats, naturalness and species diversity in Eurasian forest landscapes. Ecol Ind 10:1093–1101
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Weir JT, Schluter D (2004) Ice sheets promote speciation in boreal birds. Proc R Soc Lond B 271:1881–1887
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Yu J, Lü N, Yun F, Yue-Hua S (2011) Home range, population density, and habitat utilization of the Sichuan jay (Perisoreus internigrans). Chin Birds 2:94–100
Zhan X, Zheng Y, Wei F, Bruford MW, Jia C (2011) Molecular evidence for Pleistocene refugia at the eastern edge of the Tibetan Plateau. Mol Ecol 20:3014–3026
Zink RM, Slowinski JB (1995) Evidence from molecular systematics for decreased avian diversification in the Pleistocene epoch. Proc Natl Acad Sci 92:5832–5835
Acknowledgements
Open access funding provided by Uppsala University. Thanks to Yu Jing, Rebecka Lindbom, the Strandberg family, Ülo Väli and Magnus Johansson for the help in field, to Gunilla Engström and Maria Cortazar for the help in lab. Funding was provided by National Natural Science Foundation of China (NSFC: 31520103903 to SYH, KS), Olsson-Borghs stiftelse (to PH), Stiftelsen för Zoologisk forskning (To PH, JB, KS).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, K., Halvarsson, P., Fang, Y. et al. Genetic differentiation in Sichuan jay (Perisoreus internigrans) and its sibling species Siberian jay (P. infaustus). Conserv Genet 21, 319–327 (2020). https://doi.org/10.1007/s10592-020-01252-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-020-01252-y