
Abstract
Estimates of genetic diversity and phylogenetic affiliation represent an important resource for biodiversity assessment and a valuable guide to conservation and management. We have found a new population (Jawor—JW) of the common hamster Cricetus cricetus in western Poland that is remote from the nearest populations by 235–300 km. With the objective of genetically characterizing of this population, we compared it with other populations from Poland and Germany by taking into account sequences of four mitochondrial DNA genes and variation at 10 microsatellite loci. The JW population exhibited low levels of genetic diversity and allelic and haplotype richness, which likely reflects its extreme isolation. This factor, coupled with inbreeding and genetic drift, are major threats to JW. A neighbor-joining tree based on mtDNA haplotypes shows that JW clusters among samples representing the Central subgroup that is known from central Germany but that has not yet been identified in Poland. Findings presented here improve our understanding of the spread and diversification of the common hamster. We offer the following hypotheses to explain the observed pattern of mtDNA haplotype distribution: JW could be a byproduct of postglacial migrations or back-migrations from eastern refugia to the western part of Europe, or/and be a result of population and habitat fragmentation. We recommend translocation of individuals as an effective management strategy, both at the level of Central phylogeographic group and at the species level, to overcome the negative consequences of inbreeding and geographical isolation of the JW population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the escalating extinction crisis, both the range and the local abundance of many mammalian species have sharply decreased (Janzen 2001; Pekin and Pijanowski 2012). The common hamster Cricetus cricetus (L. 1758) is not an example of a “charismatic vertebrate”. Thus, it is not surprising that there has been almost no attention paid to its preservation until the early 1990s (Weinhold 2008). In the past several decades, this small rodent, which inhabits agrarian ecosystems and steppe habitats, has undergone a major decline in numbers to near-extinction in western and some central parts of its European range (e.g., Nechay 2000; Neumann et al. 2004, 2005; Ziomek and Banaszek 2007; Banaszek et al. 2011; Schröder et al. 2014; La Haye et al. 2014; Reiners et al. 2014; Surov et al. 2016). Interestingly, the common hamster has shown some potential for recovery following intensive ex situ and in situ conservation actions undertaken recently (e.g., Weinhold 2004; La Haye et al. 2010; Villerney et al. 2013). These management actions involve techniques such as: protective legislation, habitat restoration, fencing, establishing of captive-breeding programs for supplementation of wild populations, translocation, and monitoring (e.g., Kayser and Stubbe 2002; Jordan 2002; Weinhold 2004, 2008; Geske 2008; Kupfernagel 2008; La Haye et al. 2010; Villerney et al. 2013; O`Brien 2015). For example, on the local scale, attempts have been made to restock a population via translocation, i.e. the release of animals taken from a source population to enhance viability of a recipient population, known as augmentation, with the goal of genetic rescue or genetic restoration (see also Weeks et al. 2011). However, the genetic properties of donor and recipient populations, as well as the genetic consequences of such translocations, have not been extensively studied.
Genetic research on the common hamster has long been of interest (e.g. Neumann et al. 2004, 2005) and has generated a compelling picture of its evolutionary relationships and possible migration flows. Current genetic structure of C. cricetus is proposed to be an effect of several phenomena, such as partial extinctions, (re-)immigration, bottleneck events, in situ survival in refugia, genetic drift, and different migration routes. The negative consequences of small population size, inbreeding, lack of gene flow, and founder effects are more pronounced in populations from western Europe compared with populations from central and eastern regions (Neumann et al. 2005).
The currently observed range of C. cricetus has resulted mainly from a postglacial westward recolonization of Europe from different eastern refugia (Neumann et al. 2005). The history of its settlement in central Europe is however complicated due to repeated range expansions during the Quaternary climate oscillations. The trace fossils indicate that the common hamster was present in Poland from the Eemian interglacial (Nadachowski 1989). The westward migration from the European steppe zone was probably via two routes: a northern route across the European plains, and a southern route to the Carpathian Basin (Neumann et al. 2005). At the beginning of the Würm glacial stage, when the presence of steppe areas increased, the surge of westward expansion began to western Europe. When the glaciers expanded, a severe range retreat occurred, but the common hamster survived in the Carpathian Basin (Jánossy 1986; Neumann et al. 2005). At the end of the Würm glaciation, and in an interglacial that began 10,000–12,000 years ago, the range expanded again to encompass central and western Europe. At that time Poland became inhabited by new haplotypes derived from eastern refugia as well as from the Carpathian Basin (Banaszek et al. 2010; Korbut and Banaszek 2016).
Cricetus cricetus comprises several mitochondrial haplogroups representing western, eastern, and southern phylogeographic lineages (Neumann et al. 2005; Banaszek et al. 2012). Representatives of the North lineage in western Europe are organized into two clades: populations from western Europe (West) and populations from the central regions of Germany (including Saxony-Anhalt and Thuringia) (Central), harboring multiple mtDNA haplotypes (Neumann et al. 2004, 2005; Schröder et al. 2014). However, many aspects of the phylogenetic relationships are still unresolved, concerning for example the branching pattern in the eastern part of the species range. One group, denoted as E, spans several lineages with a western distribution border in central Europe; a Pannonia group is restricted to the Carpathian Basin and the southern part of Poland (Banaszek et al. 2010).
In Poland, individuals representing two lineages have been described to date: P3, which clusters inside the Pannonia group, and E1, one of a number of E lineages, whose western border lies in eastern Poland. This latter lineage is also present in the neighboring Ukraine from where it probably originates (Neumann et al. 2005; Banaszek et al. 2010). The P3 populations inhabited Polish areas from source populations in Moravia and Slovakia using the natural route through the Moravian Gate valley between the Sudetes and the western Carpathian Mountains in the Czech Republic (Ziomek and Banaszek 2007; Banaszek et al. 2009, 2010, 2011). In Poland, individuals representing these two lineages are found in close proximity, but the central part of Małopolska Upland creates an effective topographic and ecological barrier between them (Banaszek et al. 2012). In the majority of cases, Polish populations are geographically isolated, but some of them may have had contact with populations from western Ukraine (Korbut et al. 2013). Polish populations have diversity indices intermediate between genetically strong Pannonian and Central populations and genetically depauperate Western populations (Banaszek et al. 2011).
We focused on one particular Polish population located in the western part of the country (Jawor population, hereafter JW), representing the only Polish population remaining west of the river Oder. Geographically situated among different phylogroups (Central, Pannonia, and E1), this population may harbor unique haplotypes and may constitute a group of immigrants distinct from those already identified in Poland. Moreover, compared with populations occurring in core areas of distribution, this significantly isolated population offers an ideal model for studying levels of inbreeding and the potential loss of genetic diversity. Genetic properties of this remnant population need to be considered when making conservation decisions.
The aims of this study were (1) to define the mtDNA phylogeographic group to which JW belongs and to characterize evolutionary relationships among the populations examined representing the Pannonia, West, Central and E1 phylogroups, (2) to provide input regarding genetic parameters estimated from microsatellite data for analyzed populations, including the level of genetic diversity, structure, and effective population size, (3) using evidence from these data, to provide information useful for prioritizing the allocation of limited conservation resources in order to preserve populations of the common hamster. This information might be valuable in (1) determining a potential donor population for a translocation program, (2) future evaluation of translocation.
Materials and methods
Material and sampling
Here, we present a re-analysis of a previously published SSR data set comprising 219 individuals from Poland and Germany (Banaszek and Ziomek 2011; Banaszek et al. 2012; Reiners et al. 2011a, 2014) and 63 newly scored individuals from two Polish populations (Silesian Region) using the information provided in the ten SSR marker genotypes, Table 1, Table S1.
We sequenced four coding and non-coding mtDNA regions (in total 226 sequences) in 61 accessions, from 1 to 22 samples from each sampled field population (10) and from four samples denoted as MAM1-4, obtained from pelts of specimens collected in 2012–2015 and deposited in 2015 by collectors K. & U. Mammen in the Berlin Museum, Germany. As only a few individuals were available for each MAM sample, no SSR analysis was performed. However, the SSR analysis was completed on individuals from the closest locality in Quedlinburg (Germany), ca. 12 km E from MAM2, Table 1, Table S1.
From field populations of C. cricetus hair samples were collected from individuals using noninvasive hamster-specific hair traps in 2015 (Reiners et al. 2011a, Fig. 1; Table 1). DNA was extracted from at least ten hairs per individual using a standard Chelex 100 (Bio-Rad) protocol (Walsh et al. 1991) modified by Reiners et al. (2011b). Samples are preserved at the Department of Genetics, Adam Mickiewicz University, in Poznań, Poland.

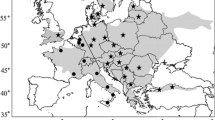
Location of sample sites throughout Poland and Germany. Samples analyzed using microsatellite markers and mtDNA are black. Populations represented by 1–4 individuals used only in mtDNA phylogenetic analysis are grey. A Polish unique JW population was indicated by a question mark. Distribution of the common hamster after Reiners et al. (2014) modified based on information stored in The Institute of Nature Conservation PAS, http://www.iop.krakow.pl/ssaki/. Table 1 provides numbers referring to sampling sites
Mitochondrial sequencing
We used four partial mitochondrial genes [control region (ctr), 16 S rRNA (16 S), cytochrome b (Cyt b), and a subunit of cytochrome c oxidase (COI)] to identify the lineages to which the analyzed individuals belong. We identified all newly scored individuals using the ctr control region. It was not possible to obtain a full data set from all accessions across all four loci. We performed DNA fragment assembly and a quality assessment with the help of SeqTrace (Stucky 2012). The final corrections were done by hand. To concatenate gene datasets, we employed Sequence Matrix v. 1.8 (Vaidya et al. 2011). For the final alignment, we trimmed sequences to the greatest length of 2072 base pairs (bp) and then used the data set to construct neighbor-joining (NJ) and maximum likelihood (ML) trees (Saitou and Nei 1987) with the help of MEGA v. 6.0. (Tamura et al. 2013).
We used the most suitable distance measure, the T92 + 0.05G distance model (Tamura 1992), for a neighbor-joining tree construction based on mtDNA sequences, and HKG + G for maximum likelihood tree construction. We chose the model using MEGA v 6.0 (Tamura et al. 2013), treating COI (1–642bp) and Cyt b (643–1380) genes as two coding regions, and 16 S (1381–1910bp) and ctr (1911–2072bp) as coding rRNAs and a non-coding DNA region, respectively; we used a gaps/missing data approach “all sites,” and the number of discrete gamma categories equal four.
A bootstrap analysis, based on 1000 replicates, compared similarities and differences between trees using TOPD/FMTS (Puigbò et al. 2007). We performed a translation into amino acid sequences using MEGA v. 6.0 to check for stop codons and non-functional coding sequences and to identify amino acid substitutions in protein-coding regions.
We conducted a statistical parsimony analysis with TCS v. 1.21 (Clement et al. 2000) to generate a haplotype network between mtDNA sequences. The connection limit was set to 95% as proposed by Hart and Sunday (2007). We performed a visualization of the network using tcsBU (Santos et al. 2015).
The mtDNA sequences have been deposited into the GenBank database (http://www.ncbi.nlm.nih.gov/GenBank), Table 2 using the submission tool BankIt (http://www.ncbi.nlm.nih.gov/BankIt/.
Population genetics analysis based on microsatellite markers
To characterize analyzed populations genetically, we employed a data set of 10 microsatellite markers (Ccrμ 10, 11, 12, 15, 17, 19, 20, IPK 03, 05, 06) (Neumann and Jansman 2004; Jakob and Mammen 2006). Protocols S1 presents all PCR reaction condition guidelines with primer sequences for mtDNA PCR amplification and details concerning the SSR and mtDNA analyses.
Using PowerMaker v. 3.25 (Liu and Muse 2005), we calculated average values of summary statistics for loci within each sampled population; we then used the same program to test the Hardy–Weinberg equilibrium for each locus. We employed ADZE (Allelic Diversity Analyzer) to calculate allelic richness within the sampled populations corrected for sample size (Szpiech et al. 2008). We tested a genotypic linkage disequilibrium with the Fisher`s method using a Markov chain (dememorization 10,000, batches 100, iterations per batch 5000) with the help of Genepop v. 4.2 (Raymond and Rousset 1995) and estimated the null allele frequency with the help of ML-NullFreq (Kalinowski and Taper 2006). With the help of FreeNa (http://www.montpellier.intra.fr/CBGP/software/FreeNa), we used the so-called ENA method (Chapuis and Estoup 2007) to perform F ST -refined estimation by excluding null alleles.
To identify population structure and assign individuals to populations of their origin, we employed STRUCTURE v. 2.3.4 (Pritchard et al. 2000; Falush et al. 2003). To imagine relationships between sampled populations, we undertook several distance-based approaches with the help of TREEFIT (Kalinowski 2009); details of this analysis are presented in Supplementary materials S1. We compared observed genetic distances between populations with the fitted genetic distances within the NJ tree and UPGMA using TREEFIT (Kalinowski 2009). We quantified the proportions of variation in the matrix of these distances explained by the NJ or UPGMA trees as R2, displaying the resulting NJ tree in TreeView v. 1.6.6.
We assessed the effect of drift within each sampled population by employing pairwise F ST for each STRUCTURE cluster against all the other following standard AMOVA as in Weir and Cockerham (1984) with the help of GenPop v. 4.2 (Rousset 2008), presenting average estimates. Because gene flow mitigates the negative effect of drift in small populations by restoring genetic variation and preventing inbreeding, we assessed recent migrations among the distinguished STRUCTURE groups using a Bayesian Markov chain Monte Carlo approach as implemented in BAYESASS v. 3.0 (Wilson and Rannala 2003). We visually assessed MCMC mixing and convergence using TRACER v. 1.6. (Rambaut et al. 2007). The isolation by distance pattern (IBD) was evaluated by assessing the correlation matrix between genetic distance and geographic distance using a Mantel`s test, as implemented in GenAlEx v. 6.5 (Peakall and Smouse 2006, 2012).
Approximate bayesian computation (ABC) analysis
We inferred effective population sizes of the STRUCTURE clusters based on an SSR data set using ABC as implemented in DIYABC v. 1.0.4.37 (Cornuet et al. 2008, 2010), available from http://www1.montpellier.inra.fr/CBGP/diyabc/. Beaumont et al. (2002) provide a full description of the ABC method. Figure 2 provides a graphic representation of competing scenarios. Models` formulation were based on our preliminary analysis (not shown). We confronted a broad spectrum of ABC explanatory models which differ in a level of complexity and a pattern of diversification. Based on the posterior probability distribution and a credibility interval for the parameter of interest, we chose three models (presented), that best fit to the data. Table S2 presents prior distribution of the simulated parameters (demographic, historical, and mutational) used in the ABC analysis. We tested different prior interval specifications for the mean microsatellite mutation rate across loci because these priors likely strongly influenced estimates of effective population size (see Lye et al. 2011). Supplementary materials S1 describe the remaining ABC analysis details.

Graphic representation of competing scenarios (sc.) modelled in DIYABC for Cricetus cricetus, focusing on the origin of Polish JW population and splits (sc. 1) Population split model assuming the origin of Polish and German populations (1–3, 6 and 4–5, 7–8, respectively) from currently sampled populations (3 and 5); population 2 (JW) is situated among Polish populations; (sc. 2) model assuming the origin of currently sampled populations from two unsampled ancestral populations (N9 and N10); JW population is situated among Polish populations; (sc. 3) is modified sc. 2, simulating the origin of JW among German populations. The populations can have different effective population sizes (N 1 − 10 ); the split events occurred at t1–t3 generations ago, to-time of sampling. Time is not at scale. STRUCTURE populations, see Table 1. Each scenario is assumed to be equally probable, meaning that these models equally likely explain the data. (Color figure online)
To compare the results of the ABC calculations concerning effective population size with another method we used a linkage disequilibrium method, which is powerful for small populations (Waples and Do 2010). An algorithm was implemented in LDNe (Waples and Do 2008). The Ne was calculated using the jackknife method for determination of 95% confidence intervals of Ne.
Results
Mitochondrial sequences were unambiguously aligned to 2076 base pairs (bp) comprising 530 bp (16 S), 645 bp (COI), 739 bp (Cyt b), and 162 bp (ctr). The 18 haplogroups comprised unique concatenated sequences in the entire data set of 226 sequences, under the 95% “parsimony” criterion, of which 63 positions were variable and 32 were parsimony informative. The mean base frequencies were as follows: A = 28.5%, C = 23.5%, G = 20.9%, T = 27.1%. Table 3 presents basic mtDNA statistics.
The phylogenetic analyses using NJ and ML approaches (the latter available upon request) produced congruent topologies regarding three well-supported groups: (1) those containing almost all samples from Germany and JW haplotype; (2) samples from Poland belonging to E1, and (3) samples from Poland belonging to Pannonia (Fig. 3a, Table S1). In the first group, it is interesting to discover that the JW population shares mitochondrial similarity with populations MAM1, 3 from central Germany, some 300 km away. The sample belonging to the E1 lineage appeared to be more closely related to the Central samples than the Pannonian samples. The data also lend support (bs 96%) to grouping of western populations (West) within the first group. The mean distance between analyzed 18 haplogroups is 1.05% (range from 0.0005 to 0.03), as shown in Table S3. Nevertheless, ML and NJ trees also include polytomies, which render the trees unsuitable for statistically evaluating their similarities and differences. The dataset (∼2000 bp) is inapplicable for inferring accurate phylogenetic relationships for samples from central Germany and JW, although within the Pannonia and E1 (Fig. 3a) a deep node can be resolved using only a single region (16 S rRNA). In summary, the JW sample clusters unsupported among the samples from central Germany representing the Central phylogeographic subgroup. There is a strict correspondence between the results of the NJ tree and parsimony network (Fig. 3b). The network connects JW with closely related, geographically cohesive haplotypes from central Germany (MAM1, 3) representing the Central phylogeographic group.

a Neighbor-joining tree of based on mtDNA haplogroups of the common hamster; tree is based on the Tamura 3-parameter distance measure and bootstrap method with 1000 bootstrap replicates with gap/missing data treatment—pairwise deletion. For more details, see Fig. 1 and Table 1, S1. Abbreviations E1 eastern phylogeographic group; b Haplotype network of concatenated sequences for the mitochondrial genes ctr, 16 S, Cyt b, and COI. Circle sizes correlate with haplotype frequencies, note that the frequencies are related to the number of individuals sequenced. Empty circles refer to missing intermediates. Poland: grey; German: black
A number of mtDNA haplotypes were unique to sampling locations, thus geographic clustering is observed. The most frequent haplotype 6 (41%) was noted in three sampling locations JW, MAM 1 and MAM 3, and required six unsampled haplotypes to connect to its closest Polish sampled haplotype (see also Table S1). The mtDNA haplotypes found in JW were previously published for samples originated in Germany, e.g., 16S: GenBank accession number: AJ633741.1 (Neumann et al. 2005), Cyt b: AJ633762.1 (Neumann et al. 2005), ctr: e.g., AJ550197.1 (Neumann et al. 2005) or KC953782 (Schröder et al. 2014), and COI: e.g. KC953805.1 (Schröder et al. 2014).
The alignment of coding sequences appears to be in frame, i.e. it does not contain premature and terminal stop codons or non-functional coding sequences. The lengths of amino acid sequences of Cyt b and COI genes are 245 and 214, respectively. The amino acid translations produced five unique protein sequences for Cyt b and four for COI, similar to those previously described in the GenBank database (similarity ranged from 99 to 100%). Individuals from JW and two other German populations show one unique amino-acid change (Cyt b: leucine instead of methionine or valine), as shown in Table S4.
Populations—genetic differentiation
The analysis of the population genetic structure based on SSR markers showed eight STRUCTURE clusters, one of which is JW. We found that the delta K value was highest at K = 8 and we found consistent results between runs at K = 8 (Fig. 4). The clusters identified in the run with the highest estimated probability reflect the samples’ close geographic proximity. The STRUCTURE results suggest also the presence of a small number of individuals with admixed ancestry. Based on Q-matrix of the 283 individuals studied, 92.58% had a coefficient of membership equal to or higher than 0.80 to their identified cluster. The presence of these individuals is attributable to shared ancestral polymorphism, gene flow, admixture events in the past, or unsampled populations.

a Bayesian clustering analysis, inferred from STRUCTURE and STRUCTURE HARVESTER, of the common hamster (C. cricetus) data (283 individuals/10 SSR loci) collected from Poland and Germany (see Fig. 1). Different colors indicate the assignment probability to different demes (K = 8); each individual is represented by vertical bars shaded in proportion to its ancestry; b Evanno`s Delta K values of the K values tested; c average F ST values of the K values tested. (Color figure online)
To infer relationships between field populations (10), we also measured and graphically displayed pairwise genetic distances using a clustering algorithm on NJ. The highest average value of R2 (R2 = 0.978) was for the NJ tree based on Theta (θ) (see Table S5, Fig. 5). The NJ tree derived from corrected genotype data for null alleles shares the same tree topology (not shown). The NJ tree showed two distinct groups (Fig. 5) clearly associated with their geographical distribution and partially reflecting the samples’ phylogeographic affinities. The first group comprises samples originating from Poland and representing Pannonia, E1 and the JW population, whereas the second group contains samples from Germany that are considered the West and Central subgroups. Incongruences are visible between relationship hypotheses regarding the origin of Polish populations based on two types of markers (mtDNA and SSR).

Populations—genetic diversity
We calculated genetic summary statistics for eight STRUCTURE clusters (Table 4). The clusters appeared highly correlated with the sampling locations. Mean He values over all loci varied between 0.346 (JW) and 0.743 (QL) with an average of 0.587. Observed heterozygosity (Ho) was lower than He in all clusters, as it was extremely low in JW. All loci were in H–W disequilibrium in the JW and JRZ clusters: nine loci in FL and MKK; six in KL; four in BOR, OP, and in a cluster composed of RAD and SZ, and two in QL. Nine pairs of loci across all clusters (20%) showed highly significant linkage disequilibrium and thus signs of hidden genetic structure. We found a consistent pattern in the occurrence of null alleles across loci for three STRUCTURE clusters (1) JRZ, (2) FL and MKK, and (3) RAD and SZ, indicating a Wahlund effect. Table S6 presents values of F ST estimations for each pair of populations with and without the ENA correction for null alleles. Regardless of the correction, F ST values indicate isolation between populations. The parameter F ST standardized by the maximum value, which has been shown to provide more accurate measures when polymorphism is high, indicates a strong population structure and near-fixation of different alleles within populations (F’ ST =0.842).
All analyzed STRUCTURE clusters showed moderate allelic richness values (mean A r = 4.01), but the value was the lowest for JW. The number of private alleles in JW was higher than that in the clusters representing the West subgroup, but lower than those in the other populations, Table 4.
Migrations and drift
All treatments performed based on the SSR data set indicate substantial differences among analyzed STRUCTURE clusters and a very low level of gene flow. The pairwise F ST values between a given cluster and the others varied between 0.091 and 0.296, but the highest values were observed for samples obtained from three Polish clusters (JW, JRZ and OP), indicating barriers to gene flow and the most pronounced effect of drift.
The pattern of migration rates within a few generations indirectly estimated by BAYESASS showed that the proportion of individuals originating from within each identified STRUCTURE cluster (non-migrants) varied from 89.97 to 97.82%, with the highest and lowest values found in the German populations. The general picture that emerges is that the analyzed clusters are strongly isolated (Table S7). Significant isolation by distance – based on the Mantel′s test – was found (r = 0.608, P < 0.010 from 10,000 randomizations).
Inferring effective population sizes with DIY ABC
In the first step, ABC based on the SSR data set yielded the strongest support, using both the direct and logistic approach, for scenario 2. The use of larger datasets 3 × 106 did not improve the best scenario’s probability over the use of 3 × 105 datasets. In scenario 2 the genetic STRUCTURE clusters, correlated to some extent with geographical location and characterized by different effective population sizes, were founded independently from two undetected and thus unsampled ancestral populations.
Relative posterior probabilities of investigated scenarios do not show strong conflictual differences between the logistic and direct approach. Nevertheless, when we used the logistic approach, which is more discriminant than the direct approach, the best-supported model appeared to be scenario 2 (set 1 priors), but when we applied the direct approach, the same scenario was preferred, but with the set 4 of prior values. Scenario 2’s posterior probabilities ranged from 0.81 (95% CI: 0.78–0.83) (set 3 priors) to 0.90 (0.88–0.915) (set 1 priors) using the logistic approach and from 0.44 (0.001–0.87) (set 3 of priors) to 0.50 (0.06–0.94) (set 4 of priors) using the direct approach (Table S8). The remaining scenarios, which assumed different origins of the JW population and different ancestral lineages, were poorly supported statistically regardless of the priors used.
Given that the results clearly favored scenario 2 (Table S8–9), we inferred the posterior distributions of parameters for this model only. The goodness-of-fit test of the scenario 2 parameter posterior combinations found five statistical differences between observed and simulated summary statistics (SuSt), (set 1 priors). Discrepancies in four of 116 of SuSt were found using the second set of prior values, whereas seven differences were found with the third set and only two using the fourth set of priors, Table S10. Table 5 presents results of the posterior distributions of parameters.
According to the methods of Cornuet et al. (2010), we estimated confidence in the model choice as Type I and II error rates from the 500 pseudo-observed data sets (PODs), (Table S9). Type I error was determined by calculating the number of cases in which the selected scenario did not have the highest posterior probability, while 500 PODs were accomplished under the best-supported scenario. Type II error rates were estimated as the proportion of cases in which the best scenario had the highest probability, while the 500 PODs were modeled with another scenario. Using the direct estimates and logistic approach methods, Type I error amounts to 11.6–18.3%, whereas Type II errors ranged from 0.00 to 12.9%.
Regardless of priors used, the mean effective population size of JW was the smallest among analyzed populations, estimated as 539–864 individuals (see Table 5). We obtained the largest values of Ne (∼6000–7000 individuals) for the populations from central Germany (QL, BOR) and the STRUCTURE cluster, containing Polish samples belonging to two different mtDNA lineages [RAD and SZ], (see Figs. 3a, 5). Between ABC parameter estimates of Ne and sample size p <− 0.26, we found no correlations. Using the LD method, we got undefined Ne estimates for the JW population (negative values); for the remaining STRUCTURE clusters, Ne values were one to three-fold lower than those obtained by the ABC calculations (not shown).
Accuracy and precision varied between parameters. The mean relative bias of the ABC analysis was low (Table 6). The effective population size for unsampled ancestral population N10, from which German populations diverged, appeared minimally biased when we employed the second set of priors (MRB = 1.605).
Discussion
From where did the Jawor population come?
A phylogenetic analysis of central European populations of Cricetus cricetus has been performed using data from approximately 2000 nucleotide-long sequences of the mtDNA four regions to define a (sub)group to which JW belongs. To our knowledge, this is the first report showing that isolates from Poland (JW) cluster together with samples from Germany. There are no recent reports of the presence of the common hamster in the western part of Poland, suggesting that JW is probably the last one and that contact with populations belonging to the Central lineage have been completely disrupted. The JW population was found 300 km east of the previously detected range edge of the Central subgroup in Europe.
The shared mtDNA haplotypes occur in JW as well as in samples belonging to the Central phylogeographical group. This might reflect historical events, i.e. repeated postglacial immigration of C. cricetus to the western parts of Europe from eastern refugia, thus supporting the hypothesis of Neumann et al. (2005). This hypothesis is indirectly supported by several other lines of evidence. Firstly, the reconstructed NJ tree suggests that mt haplotypes representing E1 are more closely related to the Central samples than to the Pannonian samples. Secondly, there are literary records describing the presence of hamsters on both sides of the Germany-Poland border in the nineteenth and twentieth centuries (Surdacki 1971; Weinhold 2008). Thirdly, there are also reports of hamsters crossing rivers and of the same hamster population inhabiting both banks of a given river (Banaszek et al. 2009; La Haye et al. 2012). This suggests that migration is possible even in a landscape bisected by a major European river like the Elbe or the Spree. Furthermore, the possibility of a contraflow from the central part of Germany to the western part of Poland cannot be excluded. There are reports of simultaneous surges of hamster numbers in central Germany until 1984, which may have led to increased migration (Nechay 2008). Moreover, such increases in numbers may have impelled hamsters to migrate in an east-southerly direction as has been observed in the northern part of Saxony by Zimmermann (1923), after Stubbe, Stubbe (1998). In summary, JW could constitute the last link in a chain of populations, reflecting the migration track that connected large distribution centers in both countries, but direction of migration remains unknown and requires further investigations.
An alternative explanation is that the observed haplotype distribution in JW and in the Central lineage may have arisen from habitat and population fragmentation. One combined ctr-COI-16S-Cyt b haplotype was found to be characteristic of both JW and populations located in the central part of Germany. A combined ctr-COI haplotype is considered the ancestral and most common haplotype of those identified in western Germany (Schröder et al. 2014 see also; Neumann et al. 2005). This finding might indicate that JW and at least some populations from those two parts of the species range descended from the same, geographically-widespread maternal progenitor (see Avise 2000). At present, a previously large, continuous area inhabited by the common hamster is shrinking in size, being divided into scattered and significantly isolated areas (Fig. 1, see e.g., Werth 1936; Weidling and Stubbe 1998; Nechay 2000; Neumann et al. 2004, 2005; Ziomek and Banaszek 2007; Tkadlec et al. 2012; Reiners et al. 2014; Korbut and Banaszek 2016). In tracking both the range expansion and its contraction, JW can be viewed as a genetic footprint of range dynamics.
Incongruences between molecular markers are commonly identified (Toews and Brelsword, 2012), as demonstrated in some C. cricetus populations (Neumann et al. (2004, 2005) and in this study. On the one hand, nuclear SSR markers showed extensive sharing of genotypes between JRZ (Pannonian) and JW, on the other hand, JW clusters among mitochondrial Central lineages. This disparity may be due to (1) differences in characteristics of molecular markers (effective population size and mutation rate), (Moore 1995; Charlesworth 1998; Hedrick 1999), or (2) long-term female philopatry and male-biased dispersal (Kayser and Stubbe 2003; Banaszek and Ziomek 2012; Banaszek et al. 2012). The extensive sharing of SSR genotypes between distant localities is expected when male-biased gene flow is common. Employing nuclear data, a significant isolation by distance based on the Mantel`s test was found (P < 0.01), which argues against male mediated gene flow, but is in accordance with the habitat and population fragmentation. Nevertheless, this pattern might be also produced in the situation when migration is constrained due to distance (Van Strien et al. 2015). Thus, analyzed SSR markers, showing clear geographic structuring, may or may not reflect the true demographic history of JW. More intensive fine-scale sampling and genotyping more loci could reveal a better resolution of the Central populations and facilitate a clearer approach to the problem of disparity in molecular markers.
Genetic characteristics of the Jawor population in comparison to neighboring populations from Poland and Germany
In the analyzed populations` set the genetic diversity value for JW was the lowest, reflecting a high degree of isolation and the lowest population size. The JW population contains only one mtDNA haplotype (6, Table S1), indicating that the population is under the strong influence of genetic drift. Through genetic drift, some alleles can disappear from a population (Wright 1931), which also may explain the low mean number of private alleles in JW. On the other hand, the almost total lack of private alleles in some German populations indicates that the positive gene flow among these populations has been disrupted relatively recently. Heterozygote deficiency was detected in all analyzed populations. The stronger deficit is in small and isolated populations rather than in larger populations occurring in close proximity to each other. This indicates non-random mating (e.g. in JW, F = 0.769, see e.g. Wright 1921). Breeding of related individuals is a consequence of restricted gene flow due to habitat fragmentation, relatively small population size, and biological constraints (e.g. Melosik et al. 2016). The common hamster does not disperse far, and its travel distance depends on sex, age, season, the location of food sources, and the incidence of reproduction outbreaks (Berdyugin and Bolshakov 1998; Nechay 2000; Kupfernagel 2008). These factors may contribute to segregation of populations into isolated reproductive units (Wahlund effect), which subsequently may lead to departures from HW proportions. Such signs of hidden genetic structure were also found in this study, but the STRUCTURE approach appeared to be insufficient to detect it, presumably due to an overwhelming effect of geographic partitioning of genetic variation (see also Evanno et al. 2005).
The results of the study are interesting from a conservation standpoint, as we provided the first single-sample effective population size estimates using a model-choice approach (ABC). Banaszek and Ziomek (2012) and Rainers et al. 2014 provided Ne estimates for the common hamster using temporal sampling. In this study, the Ne estimates ranged from a few 1000 individuals to a few 100 having the smallest value in JW (539–864 individuals). However, our estimations of Ne are at least one order of magnitude larger than those based on a single-sample linkage disequilibrium method (NeLD), (the latter not shown). The last method appeared to be sensitive, for example to persistent population fragmentation (England et al. 2010) or declines in population size (Antao et al. 2011). The model used by this method (the infinite island model under drift and migration equilibrium), requires a constant population size and migration rate over a long period of time (Wang 2005). This is probably violated in our sample due to cyclical fluctuations of population size in the common hamster and changes in the migration rates. Additional studies are required to gain better understanding of the Ne estimators using this method. Discordant results of Ne estimates depending upon the method used were also found by Leroy et al. (2013). Regardless of the method employed, several other largely unknown processes influence effective size, for example, fluctuations in size, overlapping generations, life-history traits (e.g., repeated breeders), non-random sampling (kin sampling) (Waples and Yokota 2007; Serbezov et al. 2012; Holleley et al. 2014), or sampling method used (Marucco et al. 2011). In conclusion, due to the significant variability in results depending on the method used in Ne estimations, the precise evaluation of extinction risk of JW is presently difficult (see also Franklin 1980; Soulé 1980; Gilpin and Soulé 1986; Lande 1995). It is clear, nonetheless, that JW has the smallest effective size and is subject to the heaviest inbreeding stress among the analyzed populations.
Implications for conservation
To restore genetic variability of JW and to mitigate the negative effect of inbreeding, translocations of individuals is being proposed at the level of the Central phylogeographic group as well as at the species level. Any plans of translocation should not include transfer of individuals representing different evolutionarily significant units (ESU) to avoid diminishing differently adapted variants and to prevent potentially detrimental consequences of outbreeding depression (e.g. La Haye et al. 2012, see also; Banaszek et al. 2009–2010, 2012; Neumann 2013; Schröder et al. 2014). Taking into account this position, translocation should be between JW and the Central population(s). All analyses, based on neutral genetic markers, show considerable amounts of genetic differentiation between analyzed populations, indicating uniqueness of these populations. This uniqueness most likely stems from genetic drift whose largest impact manifests in the most fragmented and isolated population. Therefore, focusing only on a few local populations as potential translocation units might further increase this fragmentation and might be ineffective in maintaining or increasing their adaptive potential in a time of progressing global warming and human pressure (Moritz 1994; Weeks et al. 2016).
Although increasing neutral genetic diversity does not guarantee that the adaptive potential of species will be protected or increased, according to Weeks et al. (2016), translocation experiments should be considered at the species level, rather than at a population, subspecies, or the ESUs levels (but see La Haye et al. 2012). If so, translocation experiments between JW and Pannonian populations should be at least preliminary assessed in captivity given that: (1) genetic similarities between the currently geographically separated populations exist, both in mitochondrial and nuclear markers, e.g. between the Pannonia and West lineages (Smulders et al. 2003) or between the Pannonia and Central lineages (Neumann et al. 2004, 2005, and results of this study); (2) reports exist showing that the contemporary hamsters have reduced fitness relative to those alive prior to the population crisis (e.g. Pucek 1981; Monecke et al. 2013), thus the beneficial effect of proposed outcrossing, such as heterosis, may be observed, and (3) biological importance of postulated ESUs is not always clearly defined and understood, and subspecies designation is not broadly accepted (Neumann et al. 2004; Banaszek et al. 2009–2010; Schröder et al. 2014). In conclusion, if the decline in genetic diversity is strictly due to neutral processes in JW, introducing a new genetic variant would be beneficial. However, the direction of translocation remains an open question until potential risks of extinction of analyzed populations and environmental sustainability are evaluated and measured (Daimler plans to build a Mercedes engine plant in Jawor). A successful outcome of this intervention would be long-term survival of the population. This would, however, depend on how quickly after translocation the effective population size reaches a minimum threshold of 1000 individuals, the number considered sufficient for maintaining its adaptive potential and evaluability (Weeks et al. 2011). Therefore, pre- and post-translocation monitoring of genetic diversity in the donors and recipient populations should be performed.
References
Antao T, Perez-Figueroa A, Luikart G (2011) Early detection of population declines: high power of genetic monitoring using effective population size estimators. Evol Appl 4:144–154
Avise JC (2000) Phylogeography: the History and Formation of Species. Harvard University Press, Cambridge, MA, p 447
Banaszek A, Ziomek J (2011) The common hamster, Cricetus cricetus (L.) populations in the Lower San River Valley. Zool Pol 56,1–4:49–58
Banaszek A, Ziomek J (2012) Genetic variation and effective population size in an isolated population of the common hamster Cricetus cricetus. Folia Zool 61(1):34–43
Banaszek A, Ziomek J, Jadwiszczak KA (2009–2010bb) Morphometric differences between the phylogeographic lineages of the common hamster Cricetus cricetus in Poland. Zool Polon 54–55(1–4):13–20
Banaszek A, Jadwiszczak KA, Ratkiewicz M, Ziomek J (2009a) Low genetic diversity and significant structuring of the common hamster populations Cricetus cricetus in Poland revealed by the mtDNA control region sequence variation. Acta Theriol 54:289–295
Banaszek A, Jadwiszczak KA, Ratkiewicz M, Ziomek J, Neumann K (2010) Population structure, colonization processes and barriers for dispersal in Polish common hamsters (Cricetus cricetus). J Zool Syst Evol Res 48(2):151–158
Banaszek A, Jadwiszczak KA, Ziomek J (2011) Genetic variability and differentiation in the Polish common hamster (Cricetus cricetus L.): genetic consequences of agricultural habitat fragmentation. Mamm Biol 76(6):665–671
Banaszek A, Ziomek J, Jadwiszczak KA, Kaczyńska E, Mirski P (2012) Identification of the barrier to gene flow between phylogeographic lineages of the common hamster Cricetus cricetus. Acta Theriol 57(3):195–204
Beaumont MA, Zhang W, Balding D (2002) Approximate Bayesian Computation in population genetics. Genetics 162:2025–2035
Berdyugin KI, Bolshakov VN (1998) The Common hamster (Cricetus cricetus L.) in the eastern part of area. In: Stubbe M, Stubbe A (eds) Őkologie und Schutz des Feldhamsters. Martin-Luther-Universität Halle-WittenbergWissenschaftliche Beiträge, Halle, pp 43–80
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24(3):621–631
Charlesworth B (1998) Measures of divergence between populations and the effect of forces that reduce variability. Mol Biol Evol 15:538–543
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cornuet JM, Santos F, Beaumont MA, Robert CP, Marin J-M, Balding DJ, Guillemaud T, Estoup A (2008) Inferring population history with DIYABC: a user friendly approach to approximate Bayesian computation. Bioinformatics 24:2713–2719
Cornuet JM, Ravigne V, Estoup A (2010) Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v 1.0). BMC Bioinform 11:401. doi:10.1186/1471-2105-11-401
England PR, Luikart G, Waples RS (2010) Early detection of population fragmentation using linkage disequilibrium estimation of effective population size. Conserv Genet 11:2425–2430
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164(4):1567–1587
Franklin IR (1980) Evolutionary change in small populations. In: Soulé ME, Wilcox BA (eds) Conservation biology: an evolutionary-ecological perspective. Sinauer Associates, Sunderland, pp 135–149
Geske Ch (2008) The Common Hamster—a species of annex IV of the habitats Directive in the German federal state of Hesse. In: Schreiber R (ed) Proceedings of the 14th Meeting of the International Hamsterworkgroup, 1–3 October 2006, Munsterschwarzach, Bayern, Germany, pp 5–9
Gilpin ME, Soulé ME (1986) Minimum viable populations: processes of species extinction. In: Soulé ME (ed) Conservation biology: the science of scarcity and diversity. Sinauer Associates, Sunderland, pp 19–34
Hart MW, Sunday J (2007) Things fall apart: biological species form unconnected parsimony networks. Biol Lett 3(5):509–512
Hedrick PW (1999) Highly variable loci and their interpretation in evolution and conservation. Evolution Int J org Evolution 53:313–318
Holleley CE, Nichols RA, Whitehead MR, Adamack AT, Gunn MR, Sherwin B (2014) Testing single-sample estimators of effective population size in genetically structured populations. Conser Genet 15(1):23–35
Jakob SS, Mammen K (2006) Eight new polymorphic microsatellite loci for genetic analyses in the endangered common hamster (Cricetus cricetus L.). Mol Ecol Notes 6(2):511–513
Jánossy D (1986) Pleistocene vertebrate faunas of Hungary. Developments in Paleontology and Stratigraphy 8. Elsevier, Amsterdam
Janzen DH (2001) Latent Extinctions – The Living Dead. In: Levin SA (ed) Encyclopedia of Biodiversity, vol 3. Academic Press, New York, pp 689–699
Jordan M (2002) Reintroduction and restocking programmes for the Common hamster. Jahrb Nassau Ver Naturkd 122:223–225
Kalinowski ST (2009) How well do evolutionary trees describe genetic relationships among populations? Heredity 102:506–513
Kalinowski ST, Taper ML (2006) Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv Genet 7:991. doi:10.1007/s10592-006-9134-9
Kayser A, Stubbe M (2002) Hamster friendly management in Germany and some aspects of habitat requirements. In: von Apeldorn RC, Stubbe M (eds) Protection of the Common hamster (Cricetus cricetus). Maastricht
Kayser A, Stubbe M (2003) Untersuchungen zum Einfluss unterschiedlicher Bewirtschaftung auf den Feldhamster Cricetus cricetus (L.) einer Leit- und Charakterart der Magdeburger Börde. Tiere im Konflikt 7. Martin-Luther-Universitäte, Halle-Wittenberg, pp 1–148
Korbut Z, Banaszek A (2016) The history of species reacting with range shifts to the oceanic-continental climate gradient in Europe. The case of the common hamster (Cricetus cricetus L.). Kosmos 6(1):69–79
Korbut Z, Rusin MY, Banaszek A (2013) The distribution of the common hamster (Cricetus cricetus) in Western Ukraine. Zool Polon 58(3–4):99–112
Kupfernagel C (2008) Crop use of the European Hamster Cricetus cricetus (L., 1758) on a hamster friendly managed study site. In: Nechay G (ed) Proceedings of the 11th Meeting of the International Hamsterworkgroup, 13–16 October 2003, Budapest, Hungary, pp 9–11
La Haye MJJ, Müskens DM, Van Kats RJM, Kuiters AT, Siepel H (2010) Agri-environmental schemes for the Common hamster (Cricetus cricetus). Why is the Dutch project successful? Asp Appl Biol 100:1–8
La Haye MJJ, Neumann K, Koelewijn HP (2012) Strong decline of gene diversity in local populations of the highly endangered Common hamster (Cricetus cricetus) in the western part of its European range. Conserv Genet 13:311–322
La Haye MJJ, Swinnen KRR, Kuiters AT, Leirs H, Siepel H (2014) Modelling population dynamics of the Common hamster (Cricetus cricetus): Timing of harvest as a critical aspect in the conservation of a highly endangered rodent. Biol Conserv 180:53–61
Lande R (1995) Mutation and conservation. Conserv Biol 9:782–791
Leroy G, Mary-Huard T, Verrier E, Danvy S, Charvolin E, Danchin-Burge C (2013) Methods to estimate effective population size using pedigree data: Examples in dog, sheep, cattle and horse. Genet Sel Evol 45:1. doi:10.1186/1297-9686-45-1
Liu K, Muse S (2005) Power Marker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Lye GC, Lepais O, Goulson D (2011) Reconstructing demographic events from population genetic data.: the introduction of bumblebees to New Zealand. Mol Ecol 20(14):2888–2900
Marucco F, Boitani L, Pletscher DH, Schwartz MK (2011) Bridging the gaps between non-invasive genetic sampling and population parameter estimation. Eur J Wild Res 57:1–13
Melosik I, Ziomek J, Winnicka K, Eichert U (2016) Genetic diversity and extinction risk in a small, declining Polish common hamster (Cricetus cricetus) population. Mamm Biol 81(6):612–622
Monecke S (2013) All things considered? Alternative reasons for hamster extinction. Zool Pol 58(3–4):41–57
Moore WS (1995) Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear gene trees. Evolution 49:718–726
Moritz C (1994) Defining “Evolutionarily Significant Units” for conservation. Tree 9:373–375
Nadachowski A (1989) Origin and history of the present rodent fauna in Poland based on fossil evidence. Acta Theriol 34:37–53
Nechay G (2000) Status of hamsters: Cricetus cricetus ,Cricetus migratorius ,Mesocricetus newtoni and other hamster species in Europe. Nature and Environment series 106. Council of Europe Publishing, Strasbourg, pp 1–73
Nechay G (2008) Peak numbers of Cricetus cricetus (L.): do they appear simultaneously? In: E Millesi, H Winkler R Hengsberger (eds) The Common hamster (Cricetus cricetus): perspectives on an endangered species. Biosyst Ecol Ser 25:69–77
Neumann K (2013) A genetic tail of two hamster species. 20th Meeting of the International Hamster Workgroup. The European hamster—new problems and prospects of their solutions. 15-17th November, Poznań, Poland, pp 13
Neumann K, Jansman H (2004) Polymorphic microsatellites for the analysis of endangered Common hamster populations (Cricetus cricetus L.). Conserv Genet 5:1127–1130
Neumann K, Jansman H, Kayser A, Maak S, Gattermann R (2004) Multiple bottleneck in threatened western European populations of the common hamster Cricetus cricetus (L.). Conserv Genet 5:181–193
Neumann K, Michaux JR, Maak S, Jansman HAH, Kayser A, Mundt G, Gattermann R (2005) Genetic spatial structure of European common hamsters (Cricetus cricetus) – a result of repeated range expansion and demographic bottlenecks. Mol Ecol 14:1473–1483
O`Brien J (2015) Saving the common hamster (Cricetus cricetus) from extinction in Alsace (France): potential flagship conservation or an exercise in futility? Hystrix 26(2):89–94
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Pekin BK, Pijanowski BC (2012) Global land use intensity and the enlargement status of mammalian species. Divers Distrib J Conserv Biogeogr 18(9):909–918
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Pucek Z (1981) Keys to vertebrates of Poland, Mammals. Polish Scientific Publishers, Warszawa, pp 1–367
Puigbò P, Garcia-Vallvé S, McInerney JO (2007) TOPD/FMTS: a new software to compare phylogenetic trees. Bioinformatics 23:1556–1558
Rambaut A, Suchard MA, Xie D, Drummond AJ (2007) Tracer v1.6, Available from http://beast.bio.ed.ac.uk.tracer
Raymond M, Rousset F (1995) GenePop (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reiners TE, Encarnação JA, Wolters V (2011a) An optimized hair trap for non-invasive genetic studies of small cryptic mammals. Eur J Wildl Res 57(4):991–995
Reiners TE, Bornmann N, Wolters V, Encarnação JA (2011b) Genetic diversity of Common hamster populations (Cricetus cricetus) revealed by non-invasive genetics. Säugetierkundl Inform 8:99–105
Reiners TE, Eidenschenk J, Neumann K, Nowak C (2014) Preservation of genetic diversity in a wild and captive population of a rapidly declining mammal, the Common hamster of the French Alsace region. Mamm Biol 79(4):240–246
Rousset F (2008) Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8(1):103–106
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Santos AM, Cabezas MP, Tavares AI, Xavier R, Branco M (2015) tcsBU: a tool to extend TCS network layout and visualization. Bioinformatics btv636. doi:10.1093/bioinformatics/btv636
Schröder O, Astrin J, Hutterer R (2014) White chest in the west: pelage colour and mitochondrial variation in the common hamster (Cricetus cricetus) across Europe. Acta Theriol 59:211–221
Serbezov D, Jorde PE, Bernatchez L, Olsen EM, Vøllestad LA (2012) Short-Term Genetic Changes: Evaluating Effective Population Size Estimates in a Comprehensively Described Brown Trout (Salmo trutta). Popul Genet 191:579–592
Smulders MJM, Snoek LB, Booy G, Vosman B (2003) Complete loss of MHC genetic diversity in the Common Hamster (Cricetus cricetus) population in The Netherlands. Consequences for conservation strategies. Conserv Genet 4:441–451
Soulé ME (1980) Thresholds for survival: maintaining fitness and evolutionary potential. In: Soulé ME, Wilcox BA (eds) Conservation biology: an evolutionary ecological perspective. Sinauer Associates, Sunderland, pp 151–169
Stubbe M, Stubbe A, Weidling A (1998) The European hamster in view of public relation and authorities, strategies of extermination and nature protection. In: Stubbe M, Stubbe A (eds) Őkologie und Schutz des Feldhamsters. Martin-Luther-Universität Halle-Wittenberg Wissenschaftliche Beiträge, Halle, pp 333–416
Stucky BJ (2012) SeqTrace: a graphical tool for rapidly processing DNA sequencing chromatograms. J Biomol Tech 23(3):90–93
Surdacki S (1971) The Distribution and Ranges of the European Hamster Cricetus cricetus (Linnaeus, 1758) in Poland. Ann UMC-S 26,12:266–284
Surov A, Banaszek A, Bogomolov P, Feoktistova N, Monecke S (2016) Dramatic global decrease in the range and the reproductive rate of the European hamster (Cricetus cricetus). Endang Species Res 31:119–145
Szpiech ZA, Jakobsson M, Rosenberg NA (2008) ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24(21):2498–2504
Tamura K (1992) Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. Mol Ecol Evol 9:678–687
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2013) MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol 30:2725–2729
Tkadlec E, Heroldovȧ M, Viśkovȧ V, BednȧřM, Zejda J (2012) Distribution of the common hamster in the Czech Republic after 2000: retreating to optimum lowland habitats. Folia Zool 61:246–253
Toews DPL, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21:3907–3930
Vaidya G, Lohman DJ, Meier R (2011) SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27(2):171–180
Van Strien MJ, Holderegger R, van Heck HJ (2015) Isolation-by-distance in landscapes: considerations for landscape genetics. Heredity 114:27–37
Villerney A, Besnard A, Grandadam J, Eidenschenck J (2013) Testing restocking methods for an endangered species: effects of predator exclusion and vegetation cover on common hamster (Cricetus cricetus) survival and reproduction. Biol Conserv 158:147–154
Walsh PS, Metzger DA, Higuchi R (1991) Chelex® 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Wang J (2005) Estimation of effective population sizes from data on genetic markers. Philos Trans R Soc Lond B Biol Sci 360:1395–1409
Waples RS, Do Ch (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Res 8:753–756
Waples RS, Do Ch (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3(3):244–262
Waples SR, Yokota M (2007) Temporal Estimates of Effective Population Size in Species With Overlapping Generations. Genetics 175(1):219–233
Weeks AR, Sgro CM, Young AG, Frankham R, Mitchell NJ, Miller KA, Byrne M, Coates DJ, Eldridge MDB, Sunnucks P, Breed MF, James EA, Hoffmann AA (2011) Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol Appl 4(6):709–725
Weeks AR, Stoklosa J, Hoffmann AA (2016) Conservation of genetic uniqueness of populations may increase extinction likelihood of endangered species: the case of Australian mammals. Front Zool 8:13–31
Weidling A, Stubbe M (1998) Actual distribution of the Common hamster (Cricetus cricetus L.) in Germany. In: Stubbe M, Stubbe A (eds) Őkologie und Schutz des Feldhamsters. Martin-Luther-Universität Halle-Wittenberg Wissenschaftliche Beiträge, Halle, pp 183–186
Weinhold U (2004) Viability of the common hamster in Western Europe—Population decline and conservation efforts. Proceedings of the 12th International hamster workgroup, 16–18 October 2004, Strasbourg, France, pp 11–19
Weinhold U (2008) Draft European action plan for the conservation of the common hamster (Cricetus cricetus L., 1758). Convention on the conservation of European wildlife and natural habitats, 28th Meeting of the Standing Committee, Strasburg, France
Weir BS, Cockerham C (1984) Estimating F-statistics for the analysis of population structure. Evolution Int J org Evolution 38(6):1358–1370
Werth E (1936) Der gegenwärtige Stand der Hamsterfrage in Deutschland. Arbeiten aus der Biologischen Reichsanstalt für Land-und Forstwirtschaft 21:201–253
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163(3):1177–1191
Wright S (1921) System of mating II. The effects of inbreeding of the genetic composition of a population. Genetics 6:124–143
Wright S (1931) Evolution in Mendelian Populations. Genetics 16(2):97–159
Zimmermann R (1923) Ueber das Vorkommen des Hamsters Cricetus cricetus (L.) und eine Erweiterung seines Verbreitungsgebietes in Sachsen. Pallasia 1(1):9–23
Ziomek J, Banaszek A (2007) The common hamster Cricetus cricetus in 701 Poland: status and current range. Folia Zool 56:235–242
Acknowledgements
The authors wish to acknowledge the anonymous reviewers for their detailed and constructive comments that help improve the manuscript. We wish also to thank Agata Nowinka who made linguistic corrections in this paper.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Melosik, I., Ziomek, J., Winnicka, K. et al. The genetic characterization of an isolated remnant population of an endangered rodent (Cricetus cricetus L.) using comparative data: implications for conservation. Conserv Genet 18, 759–775 (2017). https://doi.org/10.1007/s10592-017-0925-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-017-0925-y