
Abstract
It is not clear how the amino acid composition of the X sites in GXSXG domain affects the properties of the lipase. To address this, saturation mutation at these sites of Penicillium expansum lipase (PEL) was carried out. After screening, a mutant L133M with significantly different activity was obtained. The activity of L133M was lower than that of wild-type PEL (WT) when the reaction time was 3–10 min. However, it was 1.61 times that of WT under long-term (48 h) catalysis. WT exhibited interfacial activation when the concentration of p-nitrophenylbutyrate was higher than 3 mM, whereas L133M could only show interfacial activation at higher micelle concentration (p-nitrophenylbutyrate concentration > 5 mM), which seemed to be slower. Further analysis showed that a new hydrophobic interaction was generated between the methionine introduced at position 133 and the lid, which made the interface activation process more difficult. However, the binding energy between the activated mutant and the substrate was smaller, showing stronger activity. This work provides some reliable clues for further understanding the complex role of GXSXG domain in enzyme catalysis.
Graphic Abstract

Similar content being viewed by others
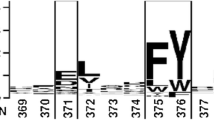

1 Introduction
Generally, lipases (EC3.1.1.3) can not only catalyze the hydrolysis of ester bond of lipid substrate [1], but also catalyze esterification, transesterification, etc. [2]. More interestingly, lipases also have catalytic promiscuity [3], and can catalyze aldol addition reactions, Michael addition [4], Knoevenagel condensation [5] and other reactions [6]. They are a kind of multifunctional biocatalysts, which can maintain their activity under a variety of reaction conditions and have potential for a wide range of uses. However, lipases are not resistant to high temperature, polar organic solvent and et al., which hinders their wide application. Therefore, it is necessary to conduct continuous and in-depth research on the relationship between the structures of the enzymes and their catalytic functions. On this basis, a variety of strategies of protein engineering can be used to modify lipase molecules directionally, so as to obtain lipase preparations with continuously improved performance [7, 8].
Penicillium expansum lipase (PEL) belongs to a large family of α/β-sheet hydrolases. Its active center is composed of a classical catalytic triad, Ser132, His241 and Asp188. When the enzyme molecule is in a closed conformation, the catalytic triad is covered with an amphipathic lid domain. At this time, the enzyme is inactive. When the lipase catalyzes the hydrolysis of oil, the enzyme molecules need to be activated at the oil–water interface, and the lid structure is opened to show catalytic activity. In our laboratory, Glu83 in the lid hinge of PEL was mutated to Lys and the mutant no longer exhibited interface activation at the oil–water interface, but it may tend to maintain an open and active structure [9]. Carriere et al. [10] interchanged the lid structures of human pancreatic lipase (HPL) and guinea pig pancreatic lipase related protein 2 (GPLRP2), resulting in HPL mutant with GPLRP2 lid domain losing original interface activation ability and its activity towards triglycerides decreased significantly. After V138 and F142 in the lid of Burkholderia cepacia lipase (BCL) were mutated into polar amino acids, the conformational change pattern of the lid was significantly changed when the mutant was activated at the interface [11]. Shu et al. [12] replaced the amino acid residues in the lid hinge of Aspergillus niger lipase and obtained a mutant Asp99Pro which can catalyze the reaction without water–oil interface. The specific activity of the mutant towards p-nitrophenyl palmitate was increased by 2.2 times. These results show that the lid and its related structural changes will significantly affect the interfacial activation and the activity.
There is also a highly conserved pentapeptide sequence (GXSXG) near the active center of PEL. In this domain, G130, S132 (one of the catalytic triad) and G134 are relatively conserved, while the amino acid composition of X131 and X133 sites shows variability among homologous lipases. Zhao et al. [13] reported that replacing H108 (equivalent to the 131st site of PEL) with tryptophan can convert MAS1 (a lipase from marine Streptomyces sp.) from non-regiospecific to sn-1,3 specific, indicating the importance of the X sites of the conserved pentapeptide sequence in defining the region specificity of MAS1. Kryukova et al. [14] described that the change of Cys173 in the GXSXG sequence of PMGL2 (a kind of mammalian hormone sensitive lipase) had a significant effect on its properties.
Changing the amino acid types of the X sites in this conserved domain of the lipase will affect the enzymatic activity to a certain extent, and may even produce new enzymatic properties. Hence, we carried out site-specific saturation mutation at positions 131/133 to explore the effect of the GXSXG domain on the catalytic performance of PEL, and to provide more data for the study of the relationship between the structure and function of the enzyme.
2 Experimental Section
2.1 Construction, Screening and Expression of Mutants
Using pGEX 4T-1-PEL as template and H131NNK/L133NNK (sense primer: ACTCTGGAAGCAGTCGGANNKTCCNNKGGTGGTGCCCTCACATCC; antisense primer: GGATGTGAGGGCACCACCMNNGGAMNNTCCGACTGCTTCCAGAGT) as primers, a whole plasmid PCR was performed to construct a mutant library. The mutant plasmids were transformed into E. coli BL21 (DE3) and the transformants were cultured on the LB plates containing ampicillin (100 µg/mL) at 37 °C for 14 h. Then, sixteen thousand transformants from LB plates were selected by transparent circle method on tributyrin emulsion agar plate containing IPTG (0.5 mM). Mutants whose hydrolytic transparency circle was larger than that of wild-type PEL were screened out and verified by DNA sequencing.
The selected mutant plasmid was digested with EcoRI and NotI and cloned into pPIC 3.5K. All the constructions were confirmed by DNA sequencing and linearized by restriction enzyme SalI and transformed into P. pastoris GS115 strain by electroporation. The transformants were grown on the MD plates at 28 °C until colonies form. The colonies were transferred to YPD plates containing 1% olive oil emulsion and 2% methanol, and cultured at 28 °C until hydrolytic transparency circles appeared around the colonies. Finally, the colony with the largest hydrolytic transparency circle was selected for expression.
The recombinant strains were incubated in 12 mL of BMGY medium at 28 °C for 24 h. The culture was transferred into BMMY medium for induction. The culture was supplemented with methanol (1%, v/v) to induce the expression of the enzyme every 24 h. After 96 h, the culture was centrifuged at 10,000×g for 15 min at 4 °C. The pellet was discarded and the supernatant was concentrated and interchanged with 10 mM Gly-NaOH buffer (pH 9.0) by ultrafiltration through a 10 kDa membrane (Millipore, USA). Enzymes in the supernatant were analyzed by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and the concentrations of the enzymes were measured using BCA Protein Assay Kit from Sangon Biotech (Shanghai, China).
2.2 Determination of the Catalytic Properties of Lipases
The esterase activity was determined using p-nitrophenyl esters as substrates [15]. Fluorescence quenching method was used to evaluate lipolytic activity using triacylglycerols with fatty acids of various chain lengths as substrates. 100 µl of solution A (20 mM Na2HPO4–NaH2PO4 solution, pH 7.6), 4 µl of solution B (8 mM fluorescein sodium solution, pH 7.6), 100 µl of dimethyl sulfoxide, 3 µl of a certain concentration of enzyme solution and 5 µl of substrate were added to a 96-well plate and mixed well. Using a multifunctional microplate reader (BioTek) at 495 nm and 37 °C, the fluorescence quenching of three parallel reactions was measured every 90 s. One unit of lipolytic activity was defined as the amount of lipase necessary to produce 1 µmol of fatty acid per minute under the above conditions.
Interfacial activation was detected using pNPB (p-nitrophenyl butyrate) in a concentration range of 0.5–7.0 mM. To detect thermostability, lipases were incubated at temperature ranging from 20 to 45 °C at 5 °C intervals for 10 min. After incubation, residual enzymatic activity was measured using pNPB as the substrate.
2.3 Gas Chromatography Analysis
The catalytic activity of wild-type lipase and mutants on olive oil was quantitatively determined by gas chromatography. Olive oil emulsion was prepared by mixing 1 vol. of olive oil and 6 vol. of 2% (w/v) polyvinyl alcohol in 50 mM glycine–NaOH buffer (pH 9.0) with a blender (10,000 rpm) until homogeneity was achieved. The reaction was divided into two groups of wild-type lipase and the mutant, with 3 replicates in each group. Every reaction system contained 270 µg wild-type PEL or mutant L133M, 2 mL olive oil emulsion, and 9 mL 20 mM PBS buffer (pH 7.4). The obtained reaction mixture was put into a shaker and reacted at 200 rpm and 37 °C. After 48 h, the reaction was terminated by adding 2 mL n-hexane, and the supernatant was collected by centrifugation. The 15 μL supernatant was transferred to thin layer chromatography (TLC) plates by sampling. After the TLC experiment was completed, part of the free fatty acids were scraped off and treated with methyl esterification, and the peak area of fatty acid methyl esters (FAMEs) in the sample was determined by gas chromatography.
Scion 436 GC (Scion instruments, CA, USA) equipped with a capillary column Agilent HP-5 (30 m × 0.25 mm × 0.25 μm; Agilent, Santa Clara, CA, USA) and a flame ionisation detector were used to analyze the FAMEs prepared above. The flow rate of carrier gas (nitrogen) was 1.1 mL/min. The initial column temperature was 145 °C and was kept for 5 min, raised to 260 °C at 4.5 °C/min, and maintained at 260 °C for 6 min. The temperatures of the injector and detector were set as 270 and 290 °C, respectively.
2.4 Interaction Analysis and Molecular Docking
Using EasyModeller software, the open-lid model of PEL was constructed with the closed-lid model of PEL (PDB:3G7N) and the open-lid model of Rhizopus oryzae lipase (PDB:4TGL) as templates. Then, open-lid models of mutant L133M was constructed based on the open-lid model of PEL, and the optimal model was selected. Autodock4.2 (The Scripps Research Institute, California, U.S.A.) was used for molecular docking between tributyrin and the model. All parameters were set to the standard values as implemented in Version 4.2. Autodock can use a genetic algorithm for the docking of small ligands into fixed protein binding sites. The docking region was defined to encompass all protein amino acids located in the hydrophobic tunnel leading to the active site of PEL. All crystallographic bound waters were removed and hydrogen atoms were added to the protein prior to docking.
3 Results and Discussion
3.1 Construction, Screening and Expression of Variants
Saturation mutation was carried out at the 131/133 position in PEL. A mutant with a hydrolysis circle significantly larger than that of wild-type lipase was screened by using the substrate (tributyrin) plates. Sequence analysis showed that the amino acid at the 133rd site of the mutant changed from Leu to Met.
In order to efficiently express mutant L133M, individual colonies of P. pastoris GS115 containing pPIC3.5K-PEL-L133M were picked from MD plates and patched on YPD plates containing 1% olive oil emulsion and 2% methanol. According to the size of the colony hydrolysis circle, the lipase-producing yeast with high expression was selected (Fig. 1).

Screening of high-expressing yeast strains. The colonies were transferred to YPD plates containing 1% olive oil emulsion and 2% methanol, and cultured at 28 °C until hydrolytic transparency circles appeared around the colonies. Finally, the colony with the largest hydrolytic transparency circle was selected for expression
Then, the expression level of wild-type PEL and its mutant L133M in Pichia pastoris was determined by SDS–PAGE. The results showed that both of them were expressed efficiently, and the purity of the lipases was higher in the fermentation broth (Fig. 2).

Production of PEL and its variant in P. pastoris. Lane M protein markers [phosphorylase b (97.2 kDa), albumin (66.4 kDa), ovalbumin (44.3 kDa), carbonic anhydrase (29 kDa), trypsin inhibitor (20.1 kDa) and lysozyme (14.3 kDa)], lane 1 supernatant of PEL/L133M, lane WT supernatant of wild-type PEL. The apparent molecular weight of the enzyme was determined by 12% (v/v) SDS–PAGE and the protein bands were visualized by Coomassie Brilliant Blue R-250
3.2 Assay of Enzymatic Properties
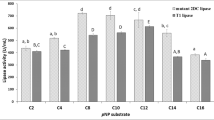
Wild-type lipase showed a preference for substrates with short-chain fatty acid in the hydrolysis reaction (for 10 min) using tributyrin, tricaprylin, olive oil as substrates, respectively (Fig. 3). However, the mutant was more inclined to catalyze the hydrolysis of tricaprylin, and the selectivity of fatty acid chain length had changed. The specific activities of wild-type toward tributyrin and tricaprylin were 14.55 U/mg and 7.56 U/mg, respectively, which were 5.64 and 1.06 times of the mutant. When the substrate was olive oil, the hydrolysis ability of wild-type and the mutant was similar and very low, and the activities were only 0.16 U/mg and 0.07 U/mg, respectively. The hydrolysis of olive oil could hardly be catalyzed in the short reaction time (10 min). The kinetic parameter kcat of wild-type PEL and the mutant reflected the same change trend (Table 1). It is noteworthy that compared with the substrate tributyrin, the km value of both wild-type PEL and the mutant towards tricaprylin decreased significantly, showing a stronger affinity.

Activity analysis of wild-type and the mutant L133M. The specific activity of WT and its mutant L133M toward triacylglycerol ester with different chain length. The results shown in this figure were averages calculated from at least three independent bioassay experiments
After 10 min of reaction, the specific activity of mutant L133M toward tributyrin was significantly lower than that of wild-type lipase, which was in contradiction with the results observed in the previous substrate (tributyrin) plate screening (48 h of reaction). However, the determination of enzyme activity is the result of a precise quantitative test, and the transparent circle method used in mutant screening is only the qualitative analysis of lipase activity. The results obtained by qualitative analysis may not be accurate. Therefore, in order to accurately determine whether the results of long-term catalytic reaction by mutant L133M and wild-type lipase are the same as those of plate screening, we used gas chromatography to determine the ability of the above two enzymes to catalyze the hydrolysis of olive oil for 48 h. The contents of free fatty acids in the reaction solution catalyzed by the two enzymes were shown in Table 2.
Using the same quality of wild-type lipase and the mutant L133M to catalyze the hydrolysis of olive oil, 3.4 mg and 5.5 mg of free fatty acids can be produced after 48 h, respectively (Table 2). The activity of the mutant L133M to catalyze the hydrolysis of olive oil was 1.6 times that of wild-type lipase, which was consistent with the results of plate screening (Fig. 1).
3.3 Interface Activation of Mutant and Wild Type Lipase
In a short period of time, the catalytic activity of wild-type lipase was higher than that of the mutant L133M, while in a longer period of time, the catalytic activity of the mutant L133M was higher than that of wild-type lipase. In order to explore what factors cause the difference in enzyme catalytic ability under different catalytic time, we first measured the thermal stability of wild-type PEL and the mutant L133M (Figs. 4, 5). The results showed that the change trend of thermal stability were similar, and there was no obvious difference. Therefore, it is unlikely that the difference between the long-term (48 h) and short-term (3–10 min) catalytic results of wild-type lipase and the mutant is due to the difference in the thermal stability of the enzymes.

Comparison of the optimum temperature between wild-type PEL and the mutant. p-Nitrophenyl palmitate, the lipase and buffer solution were mixed in a certain proportion and reacted respectively at 20 °C, 25 °C, 30 °C, 35 °C, 40 °C and 45 °C for 10 min. Then, the absorption value at 410 nm was determined. Three replicates were determined for each reaction, the average value and the relative activity was calculated. The 100% activities of the wild-type PEL (WT) and mutant L133M were 1.13 and 1.10 U/mg, respectively

Comparison of the thermostability between wild-type lipase and the mutant. Three replicates were determined for each reaction, the average value and the relative activity was calculated. The 100% activities of the wild-type PEL (WT) and mutant L133M were 0.97 and 0.89 U/mg, respectively
PEL has a “lid” structure. After this “lid” structure is activated and opened on the oil–water interface, the lipase can show its activity. In the study of interface activation, it is believed that the closing and opening of the lid is mainly related to the solubility of the substrate in the oil–water phase. When the concentration of the substrate reaches and exceeds its solubility limit, a certain concentration of insoluble substrate forms micellar aggregates in the reaction system. The existence of micellar aggregates provides ample activation power for lipases to open its lid, and the binding between enzyme and substrate is also promoted by micellar aggregates. Finally, the catalytic activity of lipase can be significantly increased [12, 16,17,18]. In the process of interface activation of lipase molecules, its specific activity will show an S-shaped curve with the change of substrate concentration. Therefore, we further explored whether there was a difference in the interface activation process between wild-type PEL and the mutant L133M (Fig. 6).

Interface activation of wild-type lipase and the mutant L133M. A the interfacial activation curve of WT and mutant L133M. Interfacial activation was detected using pNPB (p-nitrophenyl butyrate) in a concentration range of 2.0–7.0 mM. B Fluorescence Quenching curves of the catalytic products of the two enzymes with tributyrin as substrate.
The interface activation experiment was carried out at 37 °C and pH 7.6. The specific activities of wild-type PEL and mutant L133M to p-nitrophenylbutyrate (pNPB) at different concentrations (2 mM, 3 mM, 4 mM, 5 mM, 6 mM, 7 mM) were measured respectively. The results showed that wild-type lipase could show interface activation when the concentration of pNPB was > 3 mM, while the mutant L133M could show interface activation only when the concentration of substrate micelle was higher (the concentration of pNPB was higher than 5 mM) (Fig. 6a). Thus it can be seen that wild-type lipase can be activated at a lower substrate micelle concentration.
In addition, we used the fluorescence quenching method and measured the dynamic curve of the amount of products generated by the two enzymes within 10 min of the reaction in which the same concentration of tributyrin was used as the substrate (Fig. 6b). The decrease of the absorbance on the ordinate in Fig. 6b is due to the fluorescence produced by the sodium fluorescein being quenched by the fatty acids produced by the reaction. The results showed that within 1.5 min of the reaction, wild-type lipase catalyzed the production of fatty acids very quickly, and the catalytic reaction was also completed rapidly. However, mutant L133M catalyzed the production of fatty acids at a much slower rate when the reaction time was 1.5 min, and the fluorescence quenching curve declined gently. After 3 min of reaction, the rate of fatty acid production catalyzed by mutant L133M showed an obvious trend of acceleration. This showed that at the same substrate concentration, the activation process of the two enzymes was also different. At the concentration of substrate used in the experiment, wild-type lipase may be activated faster and more easily. The mutant, on the other hand, was activated more slowly. This result also supports the hypothesis that the lid structure of the mutant may be more difficult to open.
3.4 Interaction Analysis and Molecular Docking
Based on the closed-lid model of PEL (PDB: 3G7N) and the open-lid model of R. oryzae lipase (PDB: 4TGL), the closed-lid model and open-lid model of PEL and its mutant L133M were constructed by EasyModeller.
By analyzing the closed-lid structures of mutant L133M and wild-type PEL, we found that among the interactions associated with the amino acids at position 133 in the two lipase structures, only the interactions between Ile75 in the lid structure and the amino acids at position 133 were significantly different. There is a hydrophobic interaction between Met133 and Ile75 of mutant L133M, but no hydrophobic interaction between these two sites in wild-type PEL (Fig. 7). Therefore, we speculated that the switch from leucine to methionine at site 133 resulted in an additional hydrophobic interaction with Ile75 in the lid structure, which increased the resistance of the enzyme to interface activation and made it more difficult to open the lid. Therefore, during the short time (3–10 min) of enzyme activity measurement, the mutant was activated at a significantly slower speed, and it showed less activity than wild-type lipase.

Hydrophobic bond formed between the 133rd site and the amino acid in the lid of wild-type lipase or mutant L133M. A New hydrophobic interaction forming between Met133 and Ile75 in the lid of mutant L133M. B No hydrophobic interaction forming between Leu133 and Ile75 in the lid of WT. The yellow α-helix is the lid of lipase.  represents a hydrophobic interaction between residues. The complex of lipase structure and tributyrin was generated by Autodock and displayed by YASARA view
represents a hydrophobic interaction between residues. The complex of lipase structure and tributyrin was generated by Autodock and displayed by YASARA view
The open-lid models of wild-type lipase and its mutant L133M were further used with Autodock4.2 software to dock with tributyrin respectively (Fig. 8). The results showed that the binding energy of the mutant with tributyrin was − 4.09 kcal/mol, while the binding energy of wild-type lipase with tributyrin was − 3.42 kcal/mol. The lower binding energy could be correlated to the more hydrophobic nature of the substituting residue (Met) according to the Engelmen–Steitz hydrophobicity scale [19]. This indicates that the mutant may exhibit stronger substrate binding ability and higher catalytic activity compared with wild-type lipase after activation, which seems to explain why the mutant showed better catalytic effect than wild-type lipase after 48 h of reaction. Wahab et al. [20] also reported that replacing Gln at position 114 of T1 lipase with Met (rather than Leu) resulted in a greater improvement of enzyme activity.

The possible interaction models between tributyrin and PEL and its mutant L133M. A The possible interaction model between wild-type PEL and tributyrin. B The possible interaction model between the mutant L133M and tributyrin. The binding energy was shown in the figure respectively. The models were built according to the crystal structure of PEL (PDB ID: 3G7N) using autodock software package. The ligand tributyrin is shown in yellow as sticks, Ser 132 is shown in purplish red and site 133 is shown in brownish yellow. The complex of lipase structure and tributyrin was generated by Autodock and displayed by Pymol
4 Conclusions
The GXSXG domain in lipase molecules plays an important role in the molecular catalysis of the enzyme. Among them, the conserved Ser participates in the formation of the catalytic triplet, and its importance is self-evident. In addition, the less conserved X sites also affect the catalytic properties of the enzyme. In this study, saturation mutation of the X sites of PEL was carried out, and the mutant L133M with significantly changed catalytic activity was screened out. The mutant exhibited less activity than wild-type lipase during the brief 3–10 min of enzyme activity assay because of an additional hydrophobic interaction between the methionine introduced at site 133 and Ile75 in the lid structure, which made the interface activation of the mutant molecules more difficult. However, after activation (the lid opened), the binding energy between the mutant and the substrate tributyrin was smaller and the mutant showed stronger catalytic activity. Therefore, after a longer reaction time (48 h), the mutant showed better catalytic effect than wild-type lipase. These results provide some reliable experimental data for further understanding the complex role of GXSXG domain in lipase catalysis. Moreover, the selected mutant is potential to be used for the conversion of lipid substrates with greater activity.
References
Chiplunkar PP, Zhao XM, Tomke PD, Noro J, Xu B, Wang Q, Silva C, Pratap AP, Cavaco-Paulo A (2018) Ultrasound-assisted lipase catalyzed hydrolysis of aspirin methyl ester. Ultrason Sonochem 40:587–593
Xin JY, Sun LR, Chen SM, Wang Y, Xia CG (2017) Synthesis of L-ascorbyl flurbiprofenate by lipase-catalyzed esterification and transesterification reactions. Biomed Res Int 2017:1–6
Khersonsky O, Tawfik DS (2010) Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu Rev Biochem 79:471–505
Kapoor M, Gupta MN (2012) Lipase promiscuity and its biochemical applications. Process Biochem 47:555–569
Lai Y-F, Zheng H, Chai S-J, Zhang P-F, Chen X-Z (2010) Lipase-catalysed tandem Knoevenagel condensation and esterification with alcohol cosolvents. Green Chem 12:1917–1918
Sheldon RAA, Brady D, Bode MLL (2020) The Hitchhiker’s guide to biocatalysis: recent advances in the use of enzymes in organic synthesis. Chem Sci 11:2587–2605
Infanzon B, Sotelo PH, Martinez J, Diaz P (2018) Rational evolution of the unusual Y-type oxyanion hole of Rhodococcus sp CR53 lipase LipR. Enzyme Microb Technol 108:26–33
Infanzon B, Valenzuela SV, Fillat A, Javier Pastor FI, Diaz P (2014) Unusual carboxylesterase bearing a GGG(A)X-type oxyanion hole discovered in Paenibacillus barcinonensis BP-23. Biochimie 104:108–116
Tang L, Su M, Yan J, Xie S, Zhang W (2015) Lid hinge region of Penicillium expansum lipase affects enzyme activity and interfacial activation. Process Biochem 50:1218–1223
Carriere F et al (1997) Pancreatic lipase structure-function relationships by domain exchange. Biochemistry 36:239–248
Barbe S, Lafaquiere V, Guieysse D, Monsan P, Remaud-Simeon M, Andre I (2009) Insights into lid movements of Burkholderia cepacia lipase inferred from molecular dynamics simulations. Proteins Struct Funct Bioinform 77:509–523
Shu ZY, Wu JG, Xue LY, Lin RF, Jiang YM, Tang LH, Li X, Huang JZ (2011) Construction of Aspergillus niger lipase mutants with oil-water interface independence. Enzyme Microb Technol 48:129–133
Zhao Z, Hou S, Lan D, Wang X, Liu J, Khan FI, Wang Y (2017) Crystal structure of a lipase from Streptomyces sp strain W007-implications for thermostability and regiospecificity. FEBS J 284:3506–3519
Kryukova MV et al (2020) Effect of cysteine residue substitution in the GCSAG motif of the PMGL2 esterase active site on the enzyme properties. Biochemistry-Moscow 85:709–716
Tang L, Su M, Zhu L, Chi L, Zhang J, Zhou Q (2013) Substitution of Val72 residue alters the enantioselectivity and activity of Penicillium expansum lipase. World J Microbiol Biotechnol 29:145–151
Aloulou A, Rodriguez JA, Fernandez S, van Oosterhout D, Puccinelli D, Carriere F (2006) Exploring the specific features of interfacial enzymology based on lipase studies. BBA-Mol Cell Biol Lipids 1761:995–1013
Cheng M, Angkawidjaja C, Koga Y, Kanaya S (2012) Requirement of lid2 for interfacial activation of a family I.3 lipase with unique two lid structures. FEBS J 279:3727–3737
Skjold-Jorgensen J, Vind J, Svendsen A, Bjerrum MJ (2014) Altering the activation mechanism in Thermomyces lanuginosus lipase. Biochemistry 53:4152–4160
Engelman DM, Steitz TA, Goldman A (1986) Identifying non-polar transbilayer helices in amino acid sequences of membrane proteins. Annu Rev Biophys Biophys Chem 15:321–353
Wahab RA, Basri M, Rahman RNZRA et al (2012) Manipulation of the conformation and enzymatic properties of T1 lipase by site-directed mutagenesis of the protein core. Appl Biochem Biotechnol 167:612–620
Acknowledgements
This work was supported by the Grant from National Natural Science Foundation of China (81072616) and the Natural Science Foundation of Fujian province, China (2017J01442; 2020J01182).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bai, Q., Cai, Y., Yang, L. et al. Substitution of L133 with Methionine in GXSXG Domain Significantly Changed the Activity of Penicillium expansum Lipase. Catal Lett 152, 2047–2055 (2022). https://doi.org/10.1007/s10562-021-03795-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-021-03795-2