
Abstract
Isolated, coordinatively unsaturated metal sites within metal–organic framework (MOF) materials feature interesting chemical properties and offer applications as single-site catalysts. Here, we report on the recent progress in providing fundamental insight into chemical reactions occurring inside MOFs. In addition to the common form, powders, we discuss the potential of MOF thin films (SURMOFs). The combined spectroscopic and modeling approach applied to selected systems demonstrated that the catalytic activity of MOFs can be precisely tuned. A particular interesting case is the so-called defect-engineering, where structural imperfections are created in a controlled fashion. The chemical properties of MOF materials can be further modified by integration and decoration of linkers or loading with guest species, such as metal or metal–oxide nanoparticles or nanoclusters. A particularly interesting aspect of layer-by-layer approaches for the fabrication of MOF thin films is the prospect to realize tandem catalysts.
Graphical Abstract

Similar content being viewed by others

1 Introduction
Metal–organic frameworks (MOFs, also known as porous coordination polymers or PCPs) are an emerging class of porous materials of a hybrid organic/inorganic nature that combine the properties of both organic and inorganic porous materials [1,2,3,4,5,6,7]. MOFs are typically stable up to temperatures above 250 °C (in some cases, e.g., ZIF-8 [8], maximum temperatures as high as 550 °C can be tolerated). They exhibit a high degree of crystallinity and have large surface areas. The maximum degree of porosity and the size of the pores clearly exceed those of zeolites. In early 2017, the number of characterised MOFs was estimated to be 70,000 [9].
Due to these unique chemical and physical properties MOF materials have opened up new perspectives in a variety of different fields, ranging from gas storage and separation to chemical sensing, catalysis, and drug delivery [10,11,12,13,14,15,16,17,18,19,20,21]. With respect to applications in heterogeneous catalysis, different aspects must be considered. First, after adding additional coupling units (e.g., carboxylic acids or pyridine-units), molecules active in homogeneous catalysis can be incorporated into MOFs, thus providing a strategy to unite homogeneous and heterogeneous catalysis. Second, even without further functionalization, many MOFs already show interesting catalytic properties originating from the presence of coordinatively unsaturated metal sites (CUS) at the nodes. Such CUS sites are reactive for various chemical reactions. A particular advantage of such sites in MOFs is that, compared to oxide-supported metal nanoparticles (NPs), these metal cations are highly dispersed within the framework of MOFs and thus serve as isolated, “single atom” catalytic sites. Accordingly, the interest in porous MOF materials as potential catalysts (in particular single-site catalysts [22]) has intensified [22,23,24,25,26,27,28,29,30,31].
While in some MOFs CUS-sites are an intrinsic property of the MOF lattice itself, in other cases such undercoordinated sites are absent in the ideal structure and only occur as a result of defect formation within the framework material. In the case of heterogeneous catalysis, e.g., on oxide surfaces, it is well known that defect sites, such as oxygen vacancies, play a crucial role in the catalytic process [32]. Recently, it has become evident that MOFs may feature a variety of intrinsic structural defects and the corresponding (intentional) defect engineering is a powerful strategy for advanced control of MOF chemical properties (Fig. 1) [33, 34]. The defect engineered MOFs (DEMOFs), synthesized by tailoring of linkers and/or metal ions, have shown modified physical and chemical properties [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48]. Both local, isolated defects [modified CUS (mCUS)] and large-scale defects (e.g. missing nodes, mesopores, Fig. 1) can be formed in DEMOFs and these defects can have important consequences for their applications in catalysis. Furthermore, MOF materials can serve as a host matrix (support) for loading of catalytic components such as metal or metal oxide nanoparticles. These confined NPs inside MOFs may show unique properties which are significantly different from conventional supported catalysts.

Reproduced with permission from Ref. [33]. Copyright 2014 American Chemical Society
Defect-engineered MOFs (DEMOFs). The modulation of the defect structure on the micro and meso-scale by defect linker doping of the framework is shown. The blue and short red sticks represent perfect and defective linkers, respectively; the yellow and black balls represent perfect and defect metal sites, respectively; the green highlighted unit indicates the parent micropores.
In addition to MOF powders, the standard form of MOFs obtained by the conventional solvothermal synthesis, a number of methods have been developed to fabricate monolithic, crystalline, and highly oriented MOF thin films (SURMOFs) [49,50,51,52,53,54,55,56,57,58]. Among them, the kinetically controlled layer-by-layer (lbl) or liquid phase epitaxy (LPE) growth process has been extensively applied to produce well-defined SURMOFs with a high degree of crystalline order, both vertical and parallel to the substrate surface [59,60,61,62,63,64]. The highly-ordered SURMOFs not only retain the intrinsic properties of the corresponding MOFs, but also allow the design of architectures that cannot be achieved using MOF powders and to supplement their applicability as chemical sensors, smart membranes, electronic and optoelectronic devices, as well as in catalytic coatings [65,66,67,68]. More importantly, SURMOFs deposited on conducting substrates (e.g., Au) can serve as model systems for a thorough study of MOF properties by employing virtually all surface-sensitive techniques developed in Surface Science because the charging problems often hampering the analysis of bulk MOF samples are largely reduced.
Overall, the extremely high structural and compositional design ability of MOF materials holds promise for their application in catalysis, including photocatalysis and electrocatalysis. The reactive properties of MOFs can vary significantly depending on the modification of both metal centers and organic linkers as well as loading of metal/oxide NPs.
In this short report, we will highlight recent advances in the field of MOF catalytic applications, with the main emphasis being on isolated, single active sites in perfect and defect-engineered MOFs, as well as for MOF thin films and SURMOFs. We will focus on structural and electronic properties as well as chemical reactivity of the selected MOF systems based on fundamental investigations conducted primarily by our group and our collaboration partners. For other more general information about applications of MOFs in catalysis and other fields we refer the reader to numerous excellent review papers published recently (see e.g., [22, 65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82]).
We will initially discuss the chemical reactions catalyzed by perfect and defect-engineered MOFs (e.g., Cu-HKUST-1, Ru-HKUST-1) using a combined experimental and theoretical approach. This section will be followed by a brief review of the chemical nature and catalytic activity of SURMOF thin films. The next sections will focus on homochiral MOFs as well as on metal and metal–oxide NPs embedded inside MOFs. Finally, the enormous potential of SURMOF-based materials with respect to electrocatalysis will be highlighted.
2 Coordinatively Undercoordinated Single Active Sites in Pristine and Defect-Engineered MOFs (DEMOFs)
To date, MOF materials have been extensively investigated, and tremendous effort has been dedicated to their synthesis, properties and applications. However, a thorough atomic-level understanding of structural and electronic properties of the isolated, single active metal sites (CUS and mCUS) as well as the structure–activity relationship continues to be a major challenge; many crucial issues remain unanswered. This lack of information is due to the great complexity of nanostructured MOFs, especially for DEMOFs in the presence of the different types of defects described below. A comprehensive and fundamental understanding of active metal sites requires state-of-the-art analytical techniques that are suitable to probe the local chemical environments. Microscopic techniques [e.g., high-resolution transmission electron microscopy (HRTEM)] are powerful tools for structure characterization of catalysts. However, a reliable characterization of metal species in MOFs, and particularly those at defect sites in DEMOFs, is a challenge. Characterization with electron microscopy, for example, is extremely difficult since MOF materials contain organic ligands which can be easily damaged by the high energy electrons employed in TEM. Only very recently, Zhang et al. [83] have reported the high-quality atomic-resolution TEM images of UiO-66 by developing a suite of methods to overcome the experimental obstacles. Alternatively, MOFs can be characterized by spectroscopic techniques such as electron paramagnetic resonance, X-ray spectroscopy (XPS, EXAFS, XANES) and infrared spectroscopy (FTIR). Furthermore, theoretical modeling of MOFs is required to properly interpret the experimental results.
2.1 Pristine MOFs
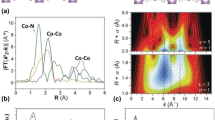
The Cu-based HKUST-1 ([Cu3btc2], btc = benzene-1,3,5-tricarboxylate) is a prototypical MOF and has been used in many studies [84]. This MOF contains so-called paddle-wheel units, where 4 carboxylate groups are bound to a Cu2-dimer. In this interesting catalytic unit, the metal ions are under-coordinated and can bind additional molecular species (e.g., CO, H2O, pyridine) at the axial position of the paddle-wheel units. Indeed, the CO oxidation reaction can be catalyzed by HKUST-1 at ambient pressure and elevated temperatures [85, 86]. In order to gain deeper insights into the active sites and reaction mechanisms, this system was systematically investigated by using high-resolution ultra-high vacuum infrared spectroscopy (UHV-FTIRS) in conjunction with density functional theory (DFT) calculations [87]. The sophisticated UHV-FTIRS apparatus combines a vacuum FTIR spectrometer (Bruker Vertex 80v) with a multi-chamber UHV system (Prevac), which not only enables in situ transmission IR experiments on MOF powders supported on an inert metal mesh, but also allows the recording of IR reflection absorption spectroscopy (IRRAS) data using a grazing incidence geometry on SURMOF thin films. This methodology has been demonstrated to be an invaluable tool to monitor chemical and photochemical reactions on the surface of metal oxide powders [32].
As shown in Fig. 2, the intrinsic Cu2+ CUS (2179 cm−1) was identified as the predominant species in HKUST-1, while a small amount of native Cu+ defects were detected as minor species (2125 cm−1, a few percent), in line with the observation for HKUST-1 thin films (see below in the section of SURMOFs) [88]. Upon exposure to O2, the UHV-FTIRS data provide direct spectroscopic evidence for a surprisingly high catalytic activity of Cu–MOFs (HKUST-1 and MOF-14, [Cu3btb2], btb = 1,2,3-benzenetrisbenzoate). Clearly, the presence of dioxygen leads to CO oxidation even at temperatures as low as 105 K. The spectroscopic information demonstrated that this reaction takes place only on the intrinsic Cu2+ CUS, whereas, rather unexpectedly, the Cu+ defect sites are inactive for the low-temperature CO oxidation. On the basis of the high-level quantum chemical calculations, a concerted mechanism was proposed, whereby the impinging O2 molecule is activated in the presence of pre-adsorbed CO and interacts simultaneously with two isocarbonyl species at neighboring CUS to yield two CO2 molecules (see Fig. 2). Notably, this mechanism differs entirely from those reported to occur in the case of CO oxidation on metal oxides or on oxide-supported metal NPs (see e.g., [89,90,91,92,93]).

Reproduced with permission from Ref. [87]. Copyright 2012 John Wiley and Sons
a UHV-FTIR spectra obtained after exposing [Cu2btc3] (HKUST-1) first to CO and subsequently to O2 at 105 K for different times (top). Intensity of the IR bands as a function of time for different CO species in the presence of molecular oxygen at 105 K (bottom). b Structure of HKUST-1. c Schematic representation of the hypothetical reaction mechanism. Starting structure: two CO molecules adsorb on opposing paddle-wheel units in the iso configuration; the O2 molecule is located (symmetrically) between these fragments (top). Resulting structure: two CO2 molecules adsorb on different paddle-wheel units (bottom).
From a catalytic point of view, isostructural MOF variants of HKUST-1 where Cu is replaced by other metals are very interesting. Such modifications, of the general type [M3(btc)2], can be obtained by incorporating Mo [94], Cr [95], or Zn [96] into the parent MOF structure. These HKUST-1 analogues possess coordinatively unsaturated metal dimers at axial positions of paddle-wheel units where adsorption and chemical reactions may take place. More challenging is the synthesis of HKUST-1 analogues where the central pair consists of metal ions in different charge states, thus yielding heterovalent paddle-wheel units. Such mixed-valence MOFs are expected to show redox activities [97, 98] and electric conducting properties [99, 100] that could be applicable to a porous electrode for batteries, fuel cells, capacitors, etc.
In contrast to all other known [M3(btc)2] frameworks that are based on M2II,II units, the Ru–MOF, [Ru3II,III(btc)2Cl1.5], exhibits mixed-valence Ru2II,III paddle-wheel units with two differently charged metal centers (Ru2+ and Ru3+), which are stabilized by additional counter ions Cl− (or in general other X− species such as OH−) to obtain an overall charge-neutral framework [101]. The combined results of UHV-FTIRS measurements using CO and CO2 as probe molecules and of accurate DFT calculations revealed that the structural and electronic properties of mixed-valence Ru–MOFs were much more complex than expected, and a straightforward assignment of the observed IR bands was impossible. Two kinds of CO species bound to Ru cation sites were identified based on the isotopic substitution and temperature-dependent IR data (Fig. 3). The C16O band at 2171 cm−1 (2120 cm−1 for C18O) was attributed to a weakly bonded CO species, whereas the low-lying C16O band at 2137 cm−1 (2085 cm−1 for C18O) originated from a CO species with an enhanced binding energy. A reasonable assignment, leading to the best-match between DFT calculations on both model systems and experimental data, is based on the assumption that one of the Cl− counter ions is transferred to a neighboring paddle-wheel, forming an anionic secondary building unit blocked by two Cl− counterions. As a consequence, the other positively charged paddle-wheel with a Ru2II,III dimer exposes two “free” CUS, where two different Ru–CO species could be formed with different frequencies and binding energies (see Fig. 3). These findings suggest that similar complexity must be taken into explicit account for many other mixed valence MOFs with intrinsically low-coordinated metal sites (CUS), which has not yet been accomplished in accurate detail.

Reproduced with permission from Ref. [101]. Copyright 2013 American Chemical Society
Temperature-dependent UHV-FTIR spectra obtained after exposing the clean Ru–MOF to a C16O and b C18O at 90 K and then elevating the temperatures. a, b, (A) exposure to C16O/ C18O at 90 K; and heated to (B) 100 K, (C) 110 K, (D) 120 K, (E) 130 K, (F) 140 K, (G) 150 K, (H) 160 K, (I) 170 K, (J) 180 K, and (K) 190 K. c Computed model systems of CO adsorption for different scenarios of Cl and CO coordinated benzoate Ru-dimer paddle wheel systems as models for local structures of Ru–MOF (atom coloring: ruthenium, green; chlorine, magenta; carbon, black; oxygen, red; hydrogen, white).
2.2 DEMOFs: Organic Linker Engineering
The above discussion of MOF catalytic properties focuses mainly on reactant adsorption and reactive transformations occurring at CUS exposed at the metal ion nodes of the framework. However, the restricted specificity and the confined coordination space of native CUS impose significant limitations, and many MOF-types (e.g., layer-pillared MOFs where Cu-paddlewheel-bound, 2D planes are stacked using, e.g., bi-pyridine units) do not contain coordinatively unsaturated sites. In analogy to heterogeneous catalysts (e.g., metal oxides and supported metal NPs), where defects are known to be the active sites in catalysis [102], it is interesting to also consider defects in MOFs. As shown in Fig. 2, reduced Cu+ species were observed to occur as intrinsic defects (with a density of a few percent) in HKUST-1.
Such defects should be absent in the perfect MOF lattice, and, indeed, more sophisticated MOF fabrication methods have been shown to reduce the concentrations of structural imperfections [103], which can act as color centers [104]. For “real” MOFs, however, the concentration of defects may be sizeable, yielding different types of unsaturated sites which strongly affect the chemical and physical properties of these porous materials [105,106,107,108,109,110,111,112]. In this context, the intentional and controlled introduction of various defects into MOF frameworks is of great importance for rational design of MOF materials with desired specific properties [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48].
A particularly elegant way to introduce defects into MOFs is by use of “defective” linkers [33, 38, 45]. By adjusting the concentration of these modified linkers relative to the regular ones the defect concentration can be precisely tuned. Such DEMOFs are of a more complex nature compared to the (more or less) “defect-free” reference materials due to the structural heterogeneity resulting from the incorporation of the defect linkers or metal ions. A comprehensive experimental characterization in conjunction with theory is required for a fundamental understanding of the defects in DEMOFs. In the case of HKUST-1 ([Cu3btc2]; Cu-BTC), Marx et al. demonstrated defect engineering of CUS via the solvothermal synthesis with carefully chosen fragmented linkers [38]. In the resulting framework, the trivalent btc3− linker was partially replaced by divalent pyridine-3,5-dicarboxylate (pydc2−). Such linker substitution was expected to yield reduced Cu+ CUS at the defect-modified paddlewheel unit. However, the expected change of the oxidation state for the copper species was not detected based on the XANES and EXAFS results in Ref. [38]. It is worth noting that the pristine HKUST-1, in the form of both powders [33, 87] and thin films (SURMOFs) [88], features intrinsically reduced Cu+ sites as the minority species, the amount of which depends on the synthesis, oxidative or reductive treatment, and the activation conditions.
Fischer and coauthors reported a series of defect-engineered HKUST-1 via systematic and controlled framework incorporation of various types of defect linkers Lx (L1-L4, see Fig. 4a) using the mixed-linker solid-solution approach as a novel synthesis strategy [33]. To gain more detailed insights into the local environments of the mCUS, UHV-FTIR spectroscopy was employed to monitor the chemisorption and thermal desorption of CO on different DEMOFs (Fig. 4). A large number of Cu-related CO bands were observed which varied in shape and intensity depending on the nature and density of the defect linkers L1–L4. These findings revealed the existence of copper species with various chemical and structural environments. The assignment of the CO vibrations was assisted by quantum mechanical/molecular mechanical (QM/MM) calculations. The combined results from UHV-FTIRS, XPS, and theory provided solid evidence for the simultaneous and controllable modification of the electronic properties and the proximate coordination space at the metal centers (mCUS) upon incorporation of defect linkers (see Fig. 4c) [33]. The doping of HKUST-1 with Lx led to the formation of reduced Cu+/Cu2+ paddlewheel units (nodes) with lowered coordination numbers. Consequently, more CO molecules can adsorb to Cu+ mCUS with a higher binding energy with respect to the parent Cu2+/Cu2+ nodes, as confirmed by temperature-dependent IR analysis [33].

Reproduced with permission from Ref. [33]. Copyright 2014 American Chemical Society
A Defect Linker concept for DEMOFs: illustration of increasing defect degree by Lx2− incorporation, i.e. btc3−/Lx2− exchange. The defect linkers L1–L3 were chosen as benzene-1,3-dicarboxylates with various functional groups at 5-position (L1: nidc2−, –NO2; L2: cydc2−; –CN; L3: hydc2−, –OH) and L4 was pyridine-3,5-dicarboxylate (pydc2−). B UHV-FTIR spectra obtained after exposing representative DEMOF samples ([Cu3(btc)2−d(Lx)d]) with different defect linkers L1–L4 to various amounts of CO at 90 K. C QM/MM computed binding modes of CO. (a–d) The QM/MM model for a local mixed valence defect Cu+/Cu2+(btc)3(pydc) being typical for sample DEMOFs (L4) is shown and energetically feasible binding modes for one to three coordinated (adsorbed) CO molecules are given (only the QM system is shown for clarity, Cu, brown; C, black; O, red; N, blue; H, white) together with computed (scaled) CO stretching normal mode frequencies (cm−1). (e) For comparison the defect is shown in a close-up, indicating the embedding of the QM system in the MM environment.
In addition to the local coordinatively undercoordinated metal sites created by integrating fragmented linkers with one coupling unit missing (type A: mCUS, see Fig. 5), lattice defects corresponding to the complete absence of metal ions (type B: node vacancies) could be formed by raising the doping concentration of defect linkers in DEMOFs. The coexistence of defects of both types A and B in Cu–DEMOFs was demonstrated by the combined UHV-FTIRS and XPS approach [113]. The node vacancies are directly related to the formation of functionalized mesopores, or large-scale defects, in DEMOFs (Fig. 1) [33]. Hupp and coworkers have reported that the introduction of node vacancies (type B defects) into HKUST-1 can finely tune the sorption properties of MOFs [114]. These results revealed the structural modulations in DEMOFs at two different length scales in a single step, which overcame restrictions of active site specificity and the confined coordination spaces at the isolated, single metal centers.

Reproduced with permission from Ref. [113]. Copyright 2017 John Wiley and Sons
Scheme of different defect types in the [M3(BTC)2] (HKUST-1, M = Cu, Ru) family as well as the regular paddlewheel units. The purple sphere in defect type B stands for a vacancy of the missing metal node.
The multivariate nature of DEMOFs represents a new dimension of tailoring functions. In catalysis, both pristine and mixed-linker Cu–MOFs showed high catalytic activity toward low-temperature hydroxylation of aromatic compounds, while the product selectivity was significantly modified in the presence of pydc2− linkers [38]. We have further investigated CO oxidation and alcohol oxidation reactions within defect-engineered HKUST-1 by IR spectroscopy. Whereas the reduced Cu+ species are inactive for the low-temperature CO oxidation, methanol oxidation occurs at mixed-valence Cu+/Cu2+ metal nodes (mCUS), indicating special catalytic properties of DEMOFs due to the coexistence of low-coordinated Cu+ CUS (i.e., more electron-rich sites) and the adjacent defect linkers (pydc2−) as functionalized groups [33].
The same kind of defect engineering has been reported for the mixed-valence [Ru3btc2] structural analogue of HKUST-1, where defects were introduced in a controlled manner by a mixed-component (native btc and defect linkers) solid-solution approach to yield Ru–DEMOFs [36, 37]. The high-level spectroscopic characterization using primarily UHV-FTIRS and X-ray based techniques (XPS, XANES, XRD) demonstrated the successful incorporation of various defect linkers that, however, did not reduce the overall integrity and robustness of the framework in a substantial way [36, 37]. Depending on the nature and concentration of fragmented linkers, both defects of type A and type B (Fig. 5) were detected. The type A defects feature reduced Ruδ+ (0 < δ < 2) sites at the modified metal nodes resulting from the fragmented or missing linkers. The formation of node vacancies (defects B) was facilitated by the doping with 5-X-isophthalic acids (5-X-ip, X = OH, H, NH2, Br) linkers where the functional groups X are much smaller than carboxylate or non-coordinating groups (e.g., H). The presence of type B defects in Ru–DEMOFs led to enhanced porosity yielding mesopores, as was observed for Cu–DEMOFs (see Fig. 1) [33].
MOF-based materials have shown great competency for the photocatalytic or electrocatalytic reduction of CO2 to CO and other value-added chemicals [115,116,117,118,119,120]. Our recent work [36, 37] revealed that the Ru–DEMOFs, synthesized by controlled incorporation of different kinds of defect linkers, exhibited unusual reactivity for the CO2 conversion to CO in a dark environment. This reaction does not occur at the “defect-free” Ru-HKUST-1, as supported by the UHV-FTIRS data (Fig. 6) which showed only CO2-related IR bands at 2335 cm−1 (12CO2) and 2272 cm−1 (13CO2). However, the introduction of pydc2− defect linkers led to the appearance of low-frequency bands at 2040 and 2000 cm−1 assigned to CO bound to modified Ruδ+ CUS, which were accompanied by a gradual decrease of CO2 signals. These findings led to the conclusion that the low-temperature (90 K) conversion of CO2 to CO is driven by strong interactions with reduced Ruδ+ CUS (i.e., more electron-rich sites) in Ru–DEMOFs. As a result, the enhanced charge transfer from Ru3d to the CO2 2πu antibonding orbital can facilitate the formation of chemisorbed CO2δ− species that may act as an intermediate to finally give rise to CO. The reactivity of Ru–DEMOFs toward the CO2 conversion depended not only on the concentration of reduced Ruδ+, but also on the nature of defect linkers. The pydc linkers further promoted the activation of CO2 due to the presence of basic pyridyl N sites in proximity to the reactive Ruδ+ mCUS (possible formation of pyridyl NOx species [121]), while the 5-OH-ip-doped Ru–DEMOFs showed much lower reactivity as compared to pydc-doped materials [37].

Reproduced with permission from Ref. [36]. Copyright 2014 John Wiley and Sons
a UHV-FTIR spectra (the regions of CO2 and CO vibrations) obtained after exposing the parent and defect-engineered Ru–MOFs to CO2 at 90 K. b Structure of the defect-engineered Ru–MOFs showing reduced and lower-coordinated Ruδ+ reactive centers and functionalized defect linkers.
Furthermore, the pydc-modified Ru–DEMOFs, pretreated with hydrogen at 423 K, exhibited dramatically enhanced activity and selectivity for olefin hydrogenation versus the competing isomerization side reaction [36]. This outcome was attributed to the efficient formation of Ru–H species via a heterolytic, base-assisted activation of dihydrogen at the cooperative active centers including reduced Ruδ+ mCUS and with the adjacent pydc2− serving as a suitable base ligand. The Ru–H species were identified by the characteristic vibrations at 1956–1975 and 2057–2076 cm−1 [36]. The proposed reaction mechanism is described in Fig. 7, where the formation of Ru–H is most likely the rate-determining step [36].

Reproduced with permission from Ref. [36]. Copyright 2014 John Wiley and Sons
Olefin hydrogenation involving base-assisted heterolytic splitting of H2 over defect-engineered Ru–MOFs. Note that the pydc linker in DEMOFs offers a basic pyridyl-N atom in the proximity of the reactive Ru centers.
The simultaneous presence of two types of defects A and B markedly affected the catalytic activities of 5-X-ip (X = OH, H, NH2, Br) engineered Ru–DEMOFs [37]. The reduced Ruδ+ sites (type A) were responsible for the enhanced catalytic performance for ethylene dimerization due to the redox properties of Ru mCUS, as observed for RhCa-X Zeolite catalysts [122]. Regarding the Paar–Knorr pyrrole synthesis, the Ru–DEMOFs showed much higher catalytic activity for the conversion of phenylamine to pyrrole, as compared to the parent Ru–MOF. Again, this transformation was attributed to the presence of type A defects, where the reduced Ruδ+ mCUS (single active sites) are bound to the O atom of the carbonyl group and facilitate nucleophilic attack by the ion-pair of the amino group of the phenylamine [37]. Interestingly, an increase in the density of incorporated 5-OH-ip led to a decrease of the yield of pyrrole [37]. This result could be explained in terms of the gradual dominance of defects B at higher doping levels; the formation of node vacancies eliminated part of the reactive Ruδ+ centers, thus accounting for the reduced catalytic activity of Ru–DEMOFs.
Overall, our results demonstrated the controlled incorporation of various defect linkers into isoreticular Cu- and Ru–MOFs (HKUST-1). The structural, electronic, and reactive properties of Cu–DEMOFs and Ru–DEMOFs varied strongly depending on the density and nature of the fragmented linkers. Other [M3(btc)2] compounds have potential to be similarly modified in a controlled manner by the choice of the functionalized defect linkers.
2.3 Mixed-Metal DEMOFs: Metal Node Engineering
Along with the linker modification in DEMOFs as discussed above, the chemical nature of MOF materials can also be precisely tuned by partial metal substitution at framework nodes. The latter approach has been employed to synthesize mixed-metal DEMOFs with the partial substitution of intrinsic Cu2+ centers in HKUST-1 by Zn2+ and other metals of 3d-row (Mn, Fe, Co) that have similar effective ionic radii and are thus closely related with Cu2+ in coordination chemistry [123, 124]. The corresponding DEMOFs showed enhanced selective sorption of O2 resulting from the incorporation of second metal ions into the framework [123]. The doping with metals of the Pd group is of special interest due to their novel catalytic activity for numerous reactions. However, the introduction of metals of the 4d or 5d row is more challenging because the presence of these metal ions makes 3D crystal formation difficult due to kinetic reasons [125, 126].
Recently, the mixed metal Pd@[Cu3−xPdx(btc)2]n with various levels of doping with Pd were obtained via one-pot synthesis [127]. The XPS results provided evidence for the simultaneous introduction of Pd2+-doped framework nodes and Pd0 NPs embedded into MOFs. As shown in Fig. 8, three Pd 3d doublets (3d5/2 and 3d3/2) were resolved in the deconvoluted Pd3d core-level spectra. The two doublets at 338.9/344.2 eV (Pd1) and 337.9/343.2 eV (Pd2) were ascribed to Pd2+ species, revealing the successful incorporation of Pd2+ into the framework of Cu-HKUST-1 leading to the formation of Cu–Pd and/or Pd–Pd paddlewheels. The doublet at 335.8/341.0 eV (Pd3) is characteristic for metallic Pd0 species; its relative concentration increased as the doping level of Pd increased, indicating the simultaneous loading of Pd0 NPs into the framework.

Reproduced with permission from Ref. [127]. Copyright 2016 Royal Society of Chemistry
a Deconvoluted XPS data of Pd@[Cu3−xPdx(btc)2]n MOFs with various doping levels of Pd in HKUST-1. b Simultaneous incorporation of Pd2+/M2+ nodes and Pd0 NPs dispersion into MOF. The Pd2+ sites in such designed MOFs play an important role in enhancing the catalytic activity of the hydrogenation of p-nitrophenol with NaBH4 to p-aminophenol.
Both Pd2+-containing MOFs [128,129,130] and Pd0@MOFs [131,132,133,134] exhibited superior catalytic performance for typical palladium-catalyzed reactions such as the Suzuki C–C coupling and hydrogenation. The Pd@[Cu3−xPdx(btc)2]n MOFs featuring both incorporated Pd2+ nodes and loaded Pd0 NPs show substantially enhanced catalytic activity toward the aqueous-phase hydrogenation of p-nitrophenol with NaBH4 to p-aminophenol, as contrasted with the pristine Cu–MOFs (HKUST-1). Furthermore, it was evident that the Pd2+ species play a key role in this reaction and are predominantly responsible for the observed high catalytic activity [127].
Along with the DEMOFs in HKUST-1 topology, intrinsic and intentionally created atomic-level defects in other types of MOFs (e.g., Zr–MOFs (UiO-66, UiO-67) [45, 47, 135,136,137,138], NU-125 [139], MOF-69 [35], MIL-53 [35]) have been the subject of numerous experimental investigations. The defects of type A (reduced metal centers with more open coordination environments) were generated either by a direct synthesis strategy or by post-synthesis approaches. In addition, Brozek and Dinca reported the fabrication of a series of mixed-metal MOF-5 analogues via isomorphous substitution at the Zn4O cornerstone (see Fig. 9), where the Zn2+ species were partially replaced by metal cations with the same (Cr2+, Mn2+, Fe2+) or different oxidation states (Ti3+, V3+, Cr3+ with Cl− as counter-ion) [140]. Furthermore, the approach of partial post-synthetic metal exchange was employed to produce mixed Al/Fe-MIL-53, Zr/Ti-UiO-66 as well as Zr/Hf-UiO-66 MOFs featuring mixed metal nodes in the framework [141]. Overall, these different types of defects have been shown to account for the high reactivity of MOF catalysts for a number of catalytic reactions [40]. The controlled incorporation of MOFs with defects of different types and concentrations represents a novel approach for the predictive rational design of MOF-based single-site catalysts at the atomic level.

Reproduced with permission from Ref. [140]. Copyright 2013 American Chemical Society
Isomorphous substitution of Zn2+ by other metal cations at the Zn4O cornerstone of MOF-5.
3 MOF Thin Films Grown by the Layer-by-Layer Method
Employing the lbl or LPE method, established by Wöll and coworkers [59], to fabricate crystalline, monolithic MOF thin films, or SURMOFs [65] affords interesting opportunities for MOF applications in catalysis. In addition to providing well-defined SURMOF substrates which can be applied for chemical transformations as observed for MOF bulk powders, the lbl method can be used to introduce different types of defects in SURMOFs, e.g. at internal interfaces in hetero-multilayer structures [65]. In addition, interstitial sites can be created by loading guest species, including metal or oxide NPs or nanoclusters (NCs), inside the parent SURMOF materials.
An instructive example are HKUST-1 SURMOFs grown on an MHDA/Au substrates (see Fig. 10). The experimental data from UHV-FTIRS and XPS were interpreted using electronic structure calculation and allowed to derive a rather consistent picture [88]. The results showed consistently the presence of a small amount (~ 4%) of reduced Cu+ species in the pristine HKUST-1 thin film. Upon heating, the temperature-induced creation of Cu+ defects was clearly observed (Fig. 10). The density of intrinsic defects varied depending on the quality of MOF thin films that could be controlled in a straightforward fashion [142]. As discussed above for MOF powders, the defect-engineered SURMOFs can be fabricated by controlled introduction of defects using different strategies such as fragmented linker incorporation [33, 38], or thermal treatment [39, 143]. It is expected that the intentional creation of defects inside SURMOFs has important consequences for tuning the electronic structure and catalytic properties of MOF materials.

Reproduced with permission from Ref. [88]. Copyright 2012 John Wiley and Sons
a Schematic drawing of HKUST-1 grown on an MHDA/Au substrate. b XP spectra of HKUST-1 SURMOF at different temperatures. c UHV-IRRAS spectra of CO adsorbed on activated HKUST-1 SURMOF at 110 K. The sample was annealed at 350 and 420 K.
The lbl method can also be used to fabricate MOF membranes. The ability to separate different molecules allows the integration of size-exclusion principles to MOF-based catalysts. The first SURMOF-based monolithic membrane was fabricated by Shekhah et al. [144] and was reported to be well-suited for the separation of small molecules, e.g., H2, N2, CO, CO2, CH4, as well as other small hydrocarbons. A more recent study has demonstrated the use of MOF-based membranes for the natural gas purification and high-value industrial separations such as butane isomers [145]. Furthermore, it has been shown that the unique opportunities to functionalize MOFs permits numerous interesting, membrane-related functionalities, including membranes where the permeability and selectivity can be switched on by light [146, 147]. As shown in Fig. 11, the photoswitchable SURMOF membrane was fabricated by assembling linkers decorated with photoresponsive azobenzene-side-groups into the framework, where the precise control of the cis/trans azobenzene ratio by controlled irradiation times with ultraviolet or visible light allows for a continuous tuning of the separation of molecular mixtures [146]. Since this approach can also be applied to fabricate large-area (larger than 20 cm × 20 cm) membranes [148], and also by applying spray-methods, it is, in principle, suited for a continuous coating process.

Reproduced with permission from Ref. [146]. Copyright 2016 Springer Nature
a Schematic illustration of tunable, remote-controllable molecular selectivity by a photoswitchable MOF membrane. b SEM cross-section image of the SURMOF membrane on the mesoporous a-Al2O3. c The structure of Cu2(AzoBPDC)2(AzoBiPyB) with the azobenzene groups. The transition between trans and cis states can be tuned by irradiation with 365 and 455 nm light, respectively.
With respect to catalysis, a particularly interesting aspect of SURMOFs is the possibility to combine two different types of chemically active MOFs into one multilayer thin film or membrane by using heteroepitaxy. This approach allows the creation of tandem catalysts where two different catalytically active components can be incorporated into one single, monolithic thin film (or membrane).
The close proximity of two different catalytically active species which can be realized by a programmed lbl approach is important in the context of reaction cascades with short-lived intermediates. In addition, a number of different catalysts may interfere, e.g., one catalyst affects the action of the other, or leads to decomposition of the second catalyst. Avoiding these unwanted effects can be achieved by anchoring the active species within a three-dimensional porous network. As opposed to just mixing the two catalysts in a liquid, embedding the two different catalysts in a MOF will maintain their individual activities.
One of the first demonstrations of such tandem catalysts realized by the lbl approach are porphyrin-based SURMOFs. In the paper by Hupp and coworkers [149], a two-component SURMOF was grown on a correspondingly functionalized substrate (see Fig. 12). This hybrid system comprising two different metallo-porphyrins (ZnMn-RPM) was shown to be active toward the epoxidation of olefin substrates (Mn-porphyrin) and for epoxide opening (Zn-porphyrin). Although this system could not yet be realized in the form of a membrane, this tandem SURMOF thin layer yielded catalytic turnover numbers which were substantially higher than the corresponding bulk MOF materials [149]. The reaction studies in this case was a tandem reaction consisting of first methoxy-styrene epoxidation catalyzed by the Mn-porphyrin to yield epoxide as the reactive intermediate. In a second step, the proximally sited Zn-porphyrin facilitates the insertion of CO2 into the epoxide, giving rise to the cyclic carbonate as the final product (Fig. 12d).

Reproduced with permission from Ref. [149]. Copyright 2016 John Wiley and Sons
a The porphyrinic dipyridine pillar used in the construction of the ZnMn-RMP MOF containing a Mn atom, which can be used as an epoxidation catalyst, and b the porphyrinic tetracarboxylic acid strut used to make the 2D sheets of the ZnMn-RPM MOF containing a Zn atom, which can act as an epoxide-opening catalyst. c Crystallographically-derived representation of a unit cell of the ZnMn-RPM framework. d Schematic representation of tandem catalysis of ZnMn-RPM for the synthesis of cyclic carbonate.
The technological impact of such tandem catalysts can be improved substantially by realizing them in the form of thin membranes through which the reactants are passed. This technology will combine size-exclusion properties with catalytic activities. A particular advantage of the molecular framework-based approach for catalyst design is that the influence of the introduction of additional side-groups to the MOF–ligands on the chemical activities can be explored in a relatively straightforward fashion. In most cases, adding additional functionalities (e.g. OH, –NH2, –CH3, etc.) to a ligand will not change the overall structure of the MOF.
This approach has been used to improve the efficiency of a UiO-66 based catalyst for phosphate ester hydrolysis, where a rate enhancement of up to 20 times was observed for UiO-66 after modification by amino (NH2) moieties that act as a proton-transfer agent during the catalysis cycle [150]. The particular advantage of MOFs in this context is the ability to integrate functional units into a porous framework material without changing the overall architecture of the MOF, which allows tuning of the chemical activity without affecting diffusivities, etc.. Additionally, the catalytic performance of MOF materials could also be tuned via the incorporation of inorganic groups containing metal centers as side-groups [22]. This procedure was termed post-synthetic metalation (PSMet) as the additional catalytically-active metal moieties can only be added post-synthetically [151]. The same decoration strategies of MOF–ligands also apply to MOF thin films, which enable the multifunctional properties of SURMOFs to be adjusted in a controlled manner (see e.g. the photoswitchable SURMOF membrane depicted in Fig. 11 [146]).
We would like to conclude this section on MOF thin films by pointing out that SURMOFs are also well-suited to study transport phenomena occurring in these framework materials in a systematic fashion. One example are so-called surface barriers, which are relevant for most porous materials. By using a quartz-crystal microbalance (QCM) based setup, Heinke et al. could demonstrate that in case of HKUST-I, these surface-barriers are not an intrinsic property of MOFs but result from surface imperfections originating from water-induced corrosion [152].
4 Homochiral MOFs and Enantioselective Asymmetric Catalysis
Kim and coworkers reported the first homochiral MOF (POST-1) that catalyzes a transesterification reaction in an enantioselective manner [153]. Since this seminal work, homochiral MOFs have been extensively investigated with the aim to rationally fabricate and engineer MOFs materials for heterogeneous asymmetric catalysis. These kinds of MOFs can be obtained via distinct strategies such as introduction of achiral active centers during synthesis, post-synthetic modification of homochiral MOFs, or incorporation of asymmetric catalysts directly into the framework [154,155,156,157,158,159,160]. The substantial potential of homochiral MOFs in enantioselective asymmetric catalytic reactions has been discussed by a number of different groups [25, 155, 161,162,163,164,165,166].
Tremendous efforts have been dedicated to homochiral MOF powder materials. However, investigations of the corresponding MOF membranes and thin films (SURMOFs) are few. Only recently, Wöll, Fischer and coworkers reported the fabrication of a series of enantiopure metal-camphorate frameworks (MCamFs) deposited on a quartz crystal microbalance (QCM) substrate via an in situ LPE lbl approach by changing the metal nodes and/or linker molecules in successive deposition cycles [167,168,169,170]. Enantioselectivity with regard to the diffusion of different enantiomers into a MOF thin film can be modulated by using linkers of different chirality [167, 168]. In addition, a thorough study of isoreticular chiral SURMOFs with identical stereogenic centers but different pore dimensions demonstrated that the pore sizes must be adjusted to achieve the highest enantioselectivity in chiral nanoporous materials [169]. Furthermore, chiral metal–organic nanoclusters (MOCs) can be loaded into the achiral MOFs, again, achieving a high selectivity between the diffusivity of different enantiomers [170]. As shown in Fig. 13, nanosized homochiral titanium oxo-clusters (Ti–MOCs) were embedded into HKUST-1 frameworks by using the LPE lbl method [170]. The resulting Ti–MOC@HKUST-1 metacrystal was quite efficient regarding enantiomer recognition and separation. Although the combination of enantiomer-selectivity with catalytically activities in MOF thin films has not been explored, we consider the potential of this direction to be enormous, in particular, when combined with membranes.

Reproduced with permission from Ref. [170]. Copyright 2016 American Chemical Society
a Structure of the R–Ti–MOC clusters. b Schematic presentation of in situ lbl growth of enantiopure Ti–MOC-loaded HKUST-1 thin film using LPE approach.
5 Metal and Oxide Nanoparticles Embedded within MOFs
In the previous paragraphs, we have demonstrated the great potential of MOFs for catalytic performance and it is far from being fully exploited. But even more possibilities exist to add further dimensions to the application of MOFs in catalysis. A particularly important one is the impregnation of MOFs with NCs or NPs. Under mild conditions (temperature below 250–550 °C, depending on type of MOF), the porous molecular frameworks are sufficiently stable to prevent sintering or agglomeration of embedded, catalytically active particles, the most severe problems encountered when exploiting the high chemical activity of such small particles for chemical transformations.
Several strategies exist with regard to loading metal, metal oxide or other chemically active NPs into the MOFs. One of the first papers in this area used metal containing precursors to realize small Pd-clusters embedded in the MOF [171]. In this case, the liberation of the metal atoms was achieved by either exposure to high pressures of H2 or by irradiation with light. It is noteworthy that the palladium clusters were substantially larger than the pores of the MOFs (Fig. 14 case b) [171]. In a similar manner, Au NPs were loaded into different MOF materials (e.g., ZIFs [172], MOF-5 [173]) and distributed homogeneously over the MOF matrix matching with the cavities (Fig. 14 case c) as confirmed by HRTEM observations. The size distribution and shape of embedded NPs was controlled by the framework structure and the functional groups at the linkers. The homogeneous distribution of confined Au NPs inside MOFs accounted for the high catalytic activity for liquid-phase aerobic oxidation of alcohols (see Fig. 14d).

Reproduced with permission from Ref. [172]. Copyright 2010 American Chemical Society
Top: three characteristic cases of microstructures for NPs supported by MOFs. a Particles typically larger than the cavity size with a preferred anchoring close to the outer surface of the MOF. b Particles evenly distributed throughout the volume of the MOF crystal but still exhibiting a broad size distribution with an average particle size exceeding the dimensions of the pores. c Particles with a narrow size distribution matching with the cavities and homogeneously distributed over the volume of MOF. Bottom: d schematic view of liquid phase alcohol oxidation with the Au@ZIF-8 material; Both benzyl alcohol (BA) and methyl benzoate (MB) are able to access the pores and can diffuse through the network.
The nano-sized bimetallic alloys are known to show enhanced catalytic performance in numerous reactions as compared to their monometallic counterparts. However, the exclusive encapsulation of bimetallic NPs with tunable compositions into MOFs is challenging [174,175,176,177,178,179]. More recently, the embedding of core–shell PdPt and RuPt nano-alloys into Zr-based MOFs (UiO-66 and its derivatives) was realized by template synthesis [180]. The resulting bimetallic core–shell NPs exhibited substrate specific size-selectivity and significantly enhanced catalytic activity for the hydrogenation of nitrobenzene compared to pure Pt loaded UiO-66 [181].
The catalytic performance of MOF materials can also be tuned in a controllable way by encapsulation of various metal oxide NPs [182,183,184,185]. The surface structure and reactivity of nanostructured ZnO particles, embedded into ZIF-8 via chemical vapor infiltration followed by oxidative annealing, were characterized by UHV-FTIRS using CO2 as a probe molecule [185]. In contrast to pure ZnO NPs exposing mainly non-polar (10–10) surfaces, the confined ZnO NPs inside ZIF-8 were dominated by polar O–ZnO and Zn–ZnO facets as well as defect sites, which were highly reactive for CO2 activation. The isolated metal oxides (e.g., ZnO, TiO2, Fe2O3) stabilized within the MOF matrix showed enhanced multifunctional (catalytic, magnetic, optical) properties and have promising applications in catalysis, photocatalysis, and other fields such as electronic devices and sensors [182,183,184,185].
In comparison to MOF bulk powders, much less information is available for the loading of MOF thin films (SURMOFs) with metal or metal oxide NPs. Recently, Wöll and coworkers reported the first fabrication of Bi2O3 NPs encapsulated into HKUST-1 thin films via a novel approach, in which bismuth-triphenyl was used to create small bismuth oxide particles into the MOF pores [186]. Also in this case the size of the largest Bi2O3 clusters slightly exceeded that of the MOF pores. The size distribution could be narrowed down substantially and at the same time shifted to lower values by adding amino groups acting as nucleation centers to the MOF linkers. Without changing the MOF architecture, lattice constant, and topology, these additional amino groups acted as nucleation centers, thus achieving a much narrower size distribution. Such bismuth oxide particles are highly active in photocatalysis, as demonstrated by the photodegradation of nuclear fast red (NFR, C14H8NO7SNa) dye [186].
6 SURMOFs and Electrocatalysis
Thin MOF films deposited on an electrode also exhibit interesting properties in electrochemistry and electrocatalysis [187, 188]. In particular, monolithic, pinhole-free SURMOFs, MOF thin films prepared using the lbl-process (see Sect. 3, above) exhibited a great potential with respect to electrochemistry [189,190,191]. Although the application of SURMOFs, both empty and after loading with electroactive compounds such as ferrocene, has been quite successful, applications in electrocatalysis have been less common.
In a recent paper by Liu, Wöll, Sun, and coworkers [192], a monolithic, pinhole-free Re-based SURMOF was grown on a conductive, transparent substrate (fluorinated tin oxide, or FTO) and exhibited well-defined electrochemical properties. As demonstrated by the X-ray diffraction (XRD) data, the MOF films were highly oriented, with the [001] direction perpendicular to the substrate. These SURMOFs were shown to be highly effective in the electrocatalytic conversion of CO2 to CO. The faradaic efficiency found in these experiments was astonishingly high, amounting to 93 ± 5%. Furthermore, the current densities which could be achieved for these high-quality monolithic coatings were found to be larger than 2 mA cm−2, thus exceeding the densities recorded for MOF thin films prepared using other methods by at least one order of magnitude.
Although few applications have so far been reported for photo-electrocatalysis of MOF thin films (e.g., MOFs as photosensitizers on TiO2 nanowires for water splitting [193]), we also foresee enormous potential in this direction. In addition to the demonstration of the suitability of SURMOFs for electrochemistry (see above), exciting properties toward photovoltaics (construction of MOF thin film based solar cells [193, 194]) have been demonstrated.
7 Summary and Outlook
The selected examples of MOF chemical activity discussed in this short review clearly demonstrate the great potential of these porous framework materials for catalysis. Active sites can be introduced into these crystalline coordination networks using a broad variety of strategies, and MOFs can be used for catalysis either as powders or in the form of thin films (SURMOFs), with the possibility to also fabricate catalytically active membranes. The combined experimental and theoretical results presented above for selected systems provided deep, fundamental insights into the structural, electronic and reactive properties of active sites in pristine and defect-engineered MOFs (DEMOFs). Depending on the system, isolated, coordinatively unsaturated metal sites (CUS) are either an intrinsic component of the perfect system or can be introduced by e.g. defect engineering or by loading with suitable guest species, thus creating the potential to fabricate single-site catalysts [22]. The catalytic performance of MOFs can be significantly enhanced by tailoring of organic linkers and/or metal cations via defect-engineering strategies in a controlled manner. The resulting DEMOFs are of highly complex nature due to the presence of different types of defects including mCUS (type A: reduced metal centers with more open coordination space) and node vacancies (type B: missing metal centers). The defects of type A, together with the proximate functionalized defect linker, were proven to play a key role in the catalytic activity of DEMOFs. The chemical and physical properties of MOF materials can be further precisely modified by adding organic or metal-containing functional groups or by embedding of metal, alloy or metal oxide NPs into frameworks.
MOF-based materials, especially MOF thin films coated on various supports, hold also great promise for the application in electrocatalysis as well as in photocatalysis and can be utilized in various environment and energy-related reactions such as water splitting and CO2 reduction.
Although the unique catalytic activities of MOF and SURMOF materials for various chemical reactions, a thorough atomic-level understanding of the active centers inside MOFs has not yet been achieved. Many important issues (e.g., the structure–activity relationships) remain to be a substantial challenge. This lack of information results from the structural complexity in pristine and defect-engineered MOFs and SURMOFs. Overall, it is mandatory to perform comprehensive fundamental studies by combining high-level microscopic (e.g. HRTEM [83]) and spectroscopic (e.g. vibrational spectroscopy and X-ray based techniques) characterizations in conjunction with theory. In this context, MOF thin films are excellent model systems which allow the application of standard methods developed in Surface Science.
References
Hoskins BF, Robson R (1990) Design and Construction of a new class of scaffolding-like materials comprising infinite polymeric frameworks of 3-D-linked molecular rods—a reappraisal of the Zn(Cn)2 and Cd(Cn)2 structures and the synthesis and structure of the diamond-related frameworks [N(CH3)4][CuIZnII(CN)4] and CuI[4,4′,4″,4‴-tetracyanotetraphenylmethane]BF4.xC6H5NO2. J Am Chem Soc 112:1546–1554
Kitagawa S, Matsuyama S, Munakata M, Emori T (1991) Synthesis and crystal structures of novel one-dimensional polymers, [{M(bpen)X}∞][M = CuI, X = PF6 –; M = AgI, X = ClO4 –; bpen = trans-1,2-bis(2-pyridyl)ethylene] and [{Cu(bpen)(CO)(CH3CN)(PF6)}∞]. J Chem Soc-Dalton Trans. https://doi.org/10.1039/DT9910002869.
Gardner GB, Venkataraman D, Moore JS, Lee S (1995) Spontaneous assembly of a hinged coordination network. Nature 374:792–795
Yaghi OM, Li HL (1995) Hydrothermal synthesis of a metal-organic framework containing large rectangular channels. J Am Chem Soc 117:10401–10402
Riou D, Ferey G (1998) Hybrid open frameworks (Mil-N). Part 3 - crystal structures of the Ht and Lt forms of MIL-7: a new vanadium propylenediphosphonate with an open-framework. influence of the synthesis temperature on the oxidation state of vanadium within the same structural type. J Mater Chem 8:2733–2735
O’Keeffe M, Eddaoudi M, Li HL, Reineke T, Yaghi OM (2000) Frameworks for extended solids: geometrical design principles. J Solid State Chem 152:3–20
Kitagawa S, Kitaura R, Noro S (2004) Functional porous coordination polymers. Angew Chem-Int Ed 43:2334–2375
Park KS, Ni Z, Cote AP, Choi JY, Huang RD, Uribe-Romo FJ, Chae HK, O’Keeffe M, Yaghi OM (2006) Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc Natl Acad Sci USA 103:10186–10191
Moghadam PZ, Li A, Wiggin SB, Tao A, Maloney AGP, Wood PA, Ward SC, Fairen-Jimenez D (2017) Development of a cambridge structural database subset: a collection of metal-organic frameworks for past, present, and future. Chem Mater 29:2618–2625
Kreno LE, Leong K, Farha OK, Allendorf M, Van Duyne RP, Hupp JT (2012) Metal-organic framework materials as chemical sensors. Chem Rev 112:1105–1125
Li JR, Sculley J, Zhou HC (2012) Metal-organic frameworks for separations. Chem Rev 112:869–932
Murray LJ, Dinca M, Long JR (2009) Hydrogen storage in metal-organic frameworks. Chem Soc Rev 38:1294–1314
Corma A, Garcia H, Xamena F (2010) Engineering metal organic frameworks for heterogeneous catalysis. Chem Rev 110:4606–4655
Ferey G (2008) Hybrid porous solids: past, present, future. Chem Soc Rev 37:191–214
Cohen SM (2012) Postsynthetic methods for the functionalization of metal-organic frameworks. Chem Rev 112:970–1000
Long JR, Yaghi OM (2009) The pervasive chemistry of metal-organic frameworks. Chem Soc Rev 38:1213–1214
Chen BL, Liang CD, Yang J, Contreras DS, Clancy YL, Lobkovsky EB, Yaghi OM, Dai S (2006) A microporous metal-organic framework for gas-chromatographic separation of alkanes. Angew Chem-Int Ed 45:1390–1393
Bloch ED, Queen WL, Krishna R, Zadrozny JM, Brown CM, Long JR (2012) Hydrocarbon separations in a metal-organic framework with open iron(II) coordination sites. Science 335:1606–1610
Talin AA, Centrone A, Ford AC, Foster ME, Stavila V, Haney P, Kinney RA, Szalai V, El Gabaly F, Yoon HP, Leonard F, Allendorf MD (2014) Tunable electrical conductivity in metal-organic framework thin-film devices. Science 343:66–69
Furukawa S, Reboul J, Diring S, Sumida K, Kitagawa S (2014) Structuring of metal-organic frameworks at the mesoscopic/macroscopic scale. Chem Soc Rev 43:5700–5734
Schneemann A, Bloch ED, Henke S, Llewellyn PL, Long JR, Fischer RA (2015) Influence of solvent-like sidechains on the adsorption of light hydrocarbons in metal-organic frameworks. Chem-Eur J 21:18764–18769
Rogge SMJ, Bavykina A, Hajek J, Garcia H, Olivos-Suarez AI, Sepulveda-Escribano A, Vimont A, Clet G, Bazin P, Kapteijn F, Daturi M, Ramos-Fernandez EV, Xamena F, Van Speybroeck V, Gascon J (2017) Metal-organic and covalent organic frameworks as single-site catalysts. Chem Soc Rev 46:3134–3184
Lee J, Farha OK, Roberts J, Scheidt KA, Nguyen ST, Hupp JT (2009) Metal-organic framework materials as catalysts. Chem Soc Rev 38:1450–1459
Farrusseng D, Aguado S, Pinel C (2009) Metal-organic frameworks: opportunities for catalysis. Angew Chem-Int Ed 48:7502–7513
Yoon M, Srirambalaji R, Kim K (2012) Homochiral metal-organic frameworks for asymmetric heterogeneous catalysis. Chem Rev 112:1196–1231
Valvekens P, Vermoortele F, De Vos D (2013) Metal-organic frameworks as catalysts: the role of metal active sites. Catal Sci Technol 3:1435–1445
Dhakshinamoorthy A, Garcia H (2014) Metal-organic frameworks as solid catalysts for the synthesis of nitrogen-containing heterocycles. Chem Soc Rev 43:5750–5765
Liu JW, Chen LF, Cui H, Zhang JY, Zhang L, Su CY (2014) Applications of metal-organic frameworks in heterogeneous supramolecular catalysis. Chem Soc Rev 43:6011–6061
Gao C, Wang J, Xu HX, Xiong YJ (2017) Coordination chemistry in the design of heterogeneous photocatalysts. Chem Soc Rev 46:2799–2823
Huang YB, Liang J, Wang XS, Cao R (2017) Multifunctional metal-organic framework catalysts: synergistic catalysis and tandem reactions. Chem Soc Rev 46:126–157
Zhu L, Liu XQ, Jiang HL, Sun LB (2017) Metal-organic frameworks for heterogeneous basic catalysis. Chem Rev 117:8129–8176
Wang YM, Wöll C (2017) IR Spectroscopic investigations of chemical and photochemical reactions on metal oxides: bridging the materials gap. Chem Soc Rev 46:1875–1932
Fang ZL, Durholt JP, Kauer M, Zhang WH, Lochenie C, Jee B, Albada B, Metzler-Nolte N, Poppl A, Weber B, Muhler M, Wang YM, Schmid R, Fischer RA (2014) Structural complexity in metal-organic frameworks: simultaneous modification of open metal sites and hierarchical porosity by systematic doping with defective linkers. J Am Chem Soc 136:9627–9636
Fang ZL, Bueken B, De Vos DE, Fischer RA (2015) Defect-engineered metal-organic frameworks. Angew Chem-Int Ed 54:7234–7254
Ravon U, Savonnet M, Aguado S, Domine ME, Janneau E, Farrusseng D (2010) Engineering of coordination polymers for shape selective alkylation of large aromatics and the role of defects. Microporous Mesoporous Mater 129:319–329
Kozachuk O, Luz I, Xamena F, Noei H, Kauer M, Albada HB, Bloch ED, Marler B, Wang YM, Muhler M, Fischer RA (2014) Multifunctional, defect-engineered metal-organic frameworks with ruthenium centers: sorption and catalytic properties. Angew Chem-Int Ed 53:7058–7062
Zhang WH, Kauer M, Halbherr O, Epp K, Guo PH, Gonzalez MI, Xiao DJ, Wiktor C, Xamena F, Wöll C, Wang YM, Muhler M, Fischer RA (2016) Ruthenium metal-organic frameworks with different defect types: influence on porosity, sorption, and catalytic properties. Chem-Eur J 22:14297–14307
Marx S, Kleist W, Baiker A (2011) Synthesis, structural properties, and catalytic behavior of Cu-BTC and mixed-linker Cu-BTC-Pydc in the oxidation of benzene derivatives. J Catal 281:76–87
Vermoortele F, Bueken B, Le Bars G, Van de Voorde B, Vandichel M, Houthoofd K, Vimont A, Daturi M, Waroquier M, Van Speybroeck V, Kirschhock C, De Vos DE (2013) Synthesis modulation as a tool to increase the catalytic activity of metal-organic frameworks: the unique case of UiO-66(Zr). J Am Chem Soc 135:11465–11468
Canivet J, Vandichel M, Farrusseng D (2016) Origin of highly active metal-organic framework catalysts: defects? defects! Dalton Trans 45:4090–4099
Liu YY, Klet RC, Hupp JT, Farha O (2016) Probing the correlations between the defects in metal-organic frameworks and their catalytic activity by an epoxide ring-opening reaction. Chem Commun 52:7806–7809
Slater B, Wang ZR, Jiang SX, Hill MR, Ladewig BP (2017) Missing linker defects in a homochiral metal-organic framework: tuning the chiral separation capacity. J Am Chem Soc 139:18322–18327
Yuan SA, Zou LF, Qin JS, Li JL, Huang L, Feng LA, Wang XA, Bosch M, Alsalme A, Cagin T, Zhou HC (2017) Construction of hierarchically porous metal-organic frameworks through linker labilization. Nat Commun 8:10
Park J, Wang ZYU, Sun LB, Chen YP, Zhou HC (2012) Introduction of functionalized mesopores to metal-organic frameworks via metal-ligand-fragment coassembly. J Am Chem Soc 134:20110–20116
Wu H, Chua YS, Krungleviciute V, Tyagi M, Chen P, Yildirim T, Zhou W (2013) Unusual and highly tunable missing-linker defects in zirconium metal-organic framework UiO-66 and their important effects on gas adsorption. J Am Chem Soc 135:10525–10532
Cliffe MJ, Wan W, Zou XD, Chater PA, Kleppe AK, Tucker MG, Wilhelm H, Funnell NP, Coudert FX, Goodwin AL (2014) Correlated defect nanoregions in a metal-organic framework. Nat Commun 5:8
Gutov OV, Hevia MG, Escudero-Adan EC, Shafir A (2015) Metal-organic framework (MOF) defects under control: insights into the missing linker sites and their implication in the reactivity of zirconium-based frameworks. Inorg Chem 54:8396–8400
Trickett CA, Gagnon KJ, Lee S, Gandara F, Burgi HB, Yaghi OM (2015) Definitive molecular level characterization of defects in UiO-66 crystals. Angew Chem-Int Ed 54:11162–11167
Kanaizuka K, Haruki R, Sakata O, Yoshimoto M, Akita Y, Kitagawa H (2008) Construction of highly oriented crystalline surface coordination polymers composed of copper dithiooxamide complexes. J Am Chem Soc 130:15778–15779
Zacher D, Shekhah O, Wöll C, Fischer RA (2009) Thin films of metal-organic frameworks. Chem Soc Rev 38:1418–1429
Shekhah O, Liu J, Fischer RA, Wöll C (2011) MOF thin films: existing and future applications. Chem Soc Rev 40:1081–1106
Betard A, Fischer RA (2012) Metal-organic framework thin films: from fundamentals to applications. Chem Rev 112:1055–1083
Bradshaw D, Garai A, Huo J (2012) Metal-organic framework growth at functional interfaces: thin films and composites for diverse applications. Chem Soc Rev 41:2344–2381
Otsubo K, Kitagawa H (2014) Metal-organic framework thin films with well-controlled growth directions confirmed by X-ray study. APL Mater 2:11
Li WJ, Tu M, Cao R, Fischer RA (2016) Metal-organic framework thin films: electrochemical fabrication techniques and corresponding applications & perspectives. J Mater Chem A 4:12356–12369
Zhuang JL, Terfort A, Wöll C (2016) Formation of oriented and patterned films of metal-organic frameworks by liquid phase epitaxy: a review. Coord Chem Rev 307:391–424
Edler KJ, Yang B (2013) Formation of mesostructured thin films at the air-liquid interface. Chem Soc Rev 42:3765–3776
Qiu SL, Xue M, Zhu GS (2014) Metal-organic framework membranes: from synthesis to separation application. Chem Soc Rev 43:6116–6140
Shekhah O, Wang H, Kowarik S, Schreiber F, Paulus M, Tolan M, Sternemann C, Evers F, Zacher D, Fischer RA, Wöll C (2007) Step-by-step route for the synthesis of metal-organic frameworks. J Am Chem Soc 129:15118–15119
Munuera C, Shekhah O, Wang H, Wöll C, Ocal C (2008) The controlled growth of oriented metal-organic frameworks on functionalized surfaces as followed by scanning force microscopy. Phys Chem Chem Phys 10:7257–7261
Shekhah O, Wang H, Zacher D, Fischer RA, Wöll C (2009) Growth mechanism of metal-organic frameworks: insights into the nucleation by employing a step-by-step route. Angew Chem-Int Ed 48:5038–5041
Shekhah O, Wang H, Paradinas M, Ocal C, Schupbach B, Terfort A, Zacher D, Fischer RA, Wöll C (2009) Controlling interpenetration in metal-organic frameworks by liquid-phase epitaxy. Nat Mater 8:481–484
Darbandi M, Arslan HK, Shekhah O, Bashir A, Birkner A, Wöll C (2010) Fabrication of free-standing ultrathin films of porous metal-organic frameworks by liquid-phase epitaxy and subsequent delamination. Phys Status Solidi-Rapid Res Lett 4:197–199
Fischer RA, Wöll C (2009) Layer-by-layer liquid-phase epitaxy of crystalline coordination polymers at surfaces. Angew Chem-Int Ed 48:6205–6208
Liu JX, Wöll C (2017) Surface-supported metal-organic framework thin films: fabrication methods, applications, and challenges. Chem Soc Rev 46:5730–5770
Falcaro P, Ricco R, Doherty CM, Liang K, Hill AJ, Styles MJ (2014) MOF positioning technology and device fabrication. Chem Soc Rev 43:5513–5560
Stavila V, Talin AA, Allendorf MD (2014) MOF-based electronic and optoelectronic devices. Chem Soc Rev 43:5994–6010
Stassen I, Burtch N, Talin A, Falcaro P, Allendorf M, Ameloot R (2017) An updated roadmap for the integration of metal-organic frameworks with electronic devices and chemical sensors. Chem Soc Rev 46:3185–3241
Yang QH, Xu Q, Jiang HL (2017) Metal-organic frameworks meet metal nanoparticles: synergistic effect for enhanced catalysis. Chem Soc Rev 46:4774–4808
Kitao T, Zhang YY, Kitagawa S, Wang B, Uemura T (2017) Hybridization of MOFs and polymers. Chem Soc Rev 46:3108–3133
Chen LY, Luque R, Li YW (2017) Controllable design of tunable nanostructures inside metal-organic frameworks. Chem Soc Rev 46:4614–4630
Medishetty R, Zareba JK, Mayer D, Samoc M, Fischer RA (2017) Nonlinear optical properties, upconversion and lasing in metal-organic frameworks. Chem Soc Rev 46:4976–5004
Li X, Liu YX, Wang J, Gascon J, Li JS, Van der Bruggen B (2017) Metal-organic frameworks based membranes for liquid separation. Chem Soc Rev 46:7124–7144
Lustig WP, Mukherjee S, Rudd ND, Desai AV, Li J, Ghosh SK (2017) Metal-organic frameworks: functional luminescent and photonic materials for sensing applications. Chem Soc Rev 46:3242–3285
Bobbitt NS, Mendonca ML, Howarth AJ, Islamoglu T, Hupp JT, Farha OK, Snurr RQ (2017) Metal-organic frameworks for the removal of toxic industrial chemicals and chemical warfare agents. Chem Soc Rev 46:3357–3385
Lian XZ, Fang Y, Joseph E, Wang Q, Li JL, Banerjee S, Lollar C, Wang X, Zhou HC (2017) Enzyme-MOF (metal-organic framework) composites. Chem Soc Rev 46:3386–3401
Adil K, Belmabkhout Y, Pillai RS, Cadiau A, Bhatt PM, Assen AH, Maurin G, Eddaoudi M (2017) Gas/vapour separation using ultra-microporous metal-organic frameworks: insights into the structure/separation relationship. Chem Soc Rev 46:3402–3430
Rubio-Martinez M, Avci-Camur C, Thornton AW, Imaz I, Maspoch D, Hill MR (2017) New synthetic routes towards MOF production at scale. Chem Soc Rev 46:3453–3480
Cao XH, Tan CL, Sindoro M, Zhang H (2017) Hybrid micro-/nano-structures derived from metal-organic frameworks: preparation and applications in energy storage and conversion. Chem Soc Rev 46:2660–2677
Easun TL, Moreau F, Yan Y, Yang SH, Schroder M (2017) Structural and dynamic studies of substrate binding in porous metal-organic frameworks. Chem Soc Rev 46:239–274
Yu JM, Xie LH, Li JR, Ma YG, Seminario JM, Balbuena PB (2017) CO2 capture and separations using MOFs: computational and experimental studies. Chem Rev 117:9674–9754
Schoedel A, Li M, Li D, O’Keeffe M, Yaghi OM (2016) Structures of metal-organic frameworks with rod secondary building units. Chem Rev 116:12466–12535
Zhang DL, Zhu YH, Liu LM, Ying XR, Hsiung CE, Sougrat R, Li K, Han Y (2018) Atomic-resolution transmission electron microscopy of electron beam-sensitive crystalline materials. Science 359:675
Chui SSY, Lo SMF, Charmant JPH, Orpen AG, Williams ID (1999) A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n. Science 283:1148–1150.
Ye JY, Liu CJ (2011) Cu-3(Btc)(2): CO oxidation over MOF based catalysts. Chem Commun 47:2167–2169
Zhao YG, Padmanabhan M, Gong QH, Tsumori N, Xu Q, Li J (2011) CO catalytic oxidation by a metal organic framework containing high density of reactive copper sites. Chem Commun 47:6377–6379
Noei H, Amirjalayer S, Muller M, Zhang XN, Schmid R, Muhler M, Fischer RA, Wang YM (2012) Low-temperature CO oxidation over Cu-based metal-organic frameworks monitored by using FTIR spectroscopy. ChemCatChem 4:755–759
St Petkov P, Vayssilov GN, Liu JX, Shekhah O, Wang YM, Wöll C, Heine T (2012) Defects in MOFs: a thorough characterization. ChemPhysChem 13:2025–2029
Haruta M, Kobayashi T, Sano H, Yamada N (1987) Novel gold catalysts for the oxidation of carbon-monoxide at a temperature far below 0-degrees-C. Chem Lett 16:405–408
Haruta A (2003) When gold is not noble: catalysis by nanoparticles. Chem Rec 3:75–87
Schubert MM, Hackenberg S, van Veen AC, Muhler M, Plzak V, Behm RJ (2001) CO oxidation over supported gold catalysts-"inert” and “active” support materials and their role for the oxygen supply during reaction. J Catal 197:113–122
Freund HJ, Meijer G, Scheffler M, Schlogl R, Wolf M (2011) CO oxidation as a prototypical reaction for heterogeneous processes. Angew Chem-Int Ed 50:10064–10094
Over H, Muhler M (2003) Catalytic Co oxidation over ruthenium—bridging the pressure gap. Prog Surf Sci 72:3–17
Kramer M, Ulrich SB, Kaskel S (2006) Synthesis and properties of the metal-organic framework Mo3(BTC)2 (TUDMOF-1). J Mater Chem 16:2245–2248.
Murray LJ, Dinca M, Yano J, Chavan S, Bordiga S, Brown CM, Long JR (2010) Highly-selective and reversible O2 binding in Cr3(1,3,5-benzenetricarboxylate)2. J Am Chem Soc 132:7856–7857.
Feldblyum JI, Liu M, Gidley DW, Matzger AJ (2011) Reconciling the discrepancies between crystallographic porosity and guest access as exemplified by Zn-HKUST-1. J Am Chem Soc 133:18257–18263
Ferey G, Millange F, Morcrette M, Serre C, Doublet ML, Greneche JM, Tarascon JM (2007) Mixed-valence Li/Fe-based metal-organic frameworks with both reversible redox and sorption properties. Angew Chem-Int Ed 46:3259–3263
Combelles C, Ben Yahia M, Pedesseau L, Doublet ML (2011) Fe-II/Fe-III mixed-valence state induced by Li-insertion into the metal-organic-framework MIL53(Fe): a DFT + U study. J Power Sources 196:3426–3432
Takaishi S, Hosoda M, Kajiwara T, Miyasaka H, Yamashita M, Nakanishi Y, Kitagawa Y, Yamaguchi K, Kobayashi A, Kitagawa H (2009) Electroconductive porous coordination polymer Cu[Cu(pdt)2] composed of donor and acceptor building units. Inorg Chem 48:9048–9050.
Kobayashi Y, Jacobs B, Allendorf MD, Long JR (2010) Conductivity, doping, and redox chemistry of a microporous dithiolene-based metal-organic framework. Chem Mater 22:4120–4122
Noei H, Kozachuk O, Amirjalayer S, Bureekaew S, Kauer M, Schmid R, Marler B, Muhler M, Fischer RA, Wang YM (2013) CO adsorption on a mixed-valence ruthenium metal-organic framework studied by UHV-FTIR spectroscopy and DFT calculations. J Phys Chem C 117:5658–5666
Xu MC, Noei H, Fink K, Muhler M, Wang YM, Wöll C (2012) The surface science approach for understanding reactions on oxide powders: the importance of IR spectroscopy. Angew Chem-Int Ed 51:4731–4734
Gu ZG, Pfriem A, Hamsch S, Breitwieser H, Wohlgemuth J, Heinke L, Gliemann H, Wöll C (2015) Transparent films of metal-organic frameworks for optical applications. Microporous Mesoporous Mater 211:82–87
Muller K, Fink K, Schottner L, Koenig M, Heinke L, Wöll C (2017) Defects as color centers: the apparent color of metal-organic frameworks containing Cu2+-based paddle-wheel units. ACS Appl Mater Interfaces 9:37463–37467.
Shoaee M, Agger JR, Anderson MW, Attfield MP (2008) Crystal form, defects and growth of the metal organic framework Hkust-1 revealed by atomic force microscopy. CrystEngComm 10:646–648
Cairns AB, Goodwin AL (2013) Structural disorder in molecular framework materials. Chem Soc Rev 42:4881–4893
Chizallet C, Lazare S, Bazer-Bachi D, Bonnier F, Lecocq V, Soyer E, Quoineaud AA, Bats N (2010) Catalysis of transesterification by a nonfunctionalized metal-organic framework: acido-basicity at the external surface of ZIF-8 probed by FTIR and Ab initio calculations. J Am Chem Soc 132:12365–12377
Xamena F, Cirujano FG, Corma A (2012) An Unexpected bifunctional acid base catalysis in Irmof-3 for Knoevenagel condensation reactions. Microporous Mesoporous Mater 157:112–117
Bunck DN, Dichtel WR (2013) Mixed linker strategies for organic framework functionalization. Chem-Eur J 19:818–827
Park TH, Hickman AJ, Koh K, Martin S, Wong-Foy AG, Sanford MS, Matzger AJ (2011) Highly dispersed palladium(II) in a defective metal-organic framework: application to C–H activation and functionalization. J Am Chem Soc 133:20138–20141
Diring S, Furukawa S, Takashima Y, Tsuruoka T, Kitagawa S (2010) Controlled multiscale synthesis of porous coordination polymer in nano/micro regimes. Chem Mater 22:4531–4538
Al-Janabi N, Fan XL, Siperstein FR (2016) Assessment of MOF’s Quality: quantifying defect content in crystalline porous materials. J Phys Chem Lett 7:1490–1494
Zhang WH, Kauer M, Guo PH, Kunze S, Cwik S, Muhler M, Wang YM, Epp K, Kieslich G, Fischer RA (2017) Impact of synthesis parameters on the formation of defects in HKUST-1. Eur J Inorg Chem 2017:925–931
Barin G, Krungleviciute V, Gutov O, Hupp JT, Yildirim T, Farha OK (2014) Defect creation by linker fragmentation in metal-organic frameworks and its effects on gas uptake properties. Inorg Chem 53:6914–6919
S. Diercks C, Liu Y, Cordova K, M. Yaghi O (2018) The role of reticular chemistry in the design of CO2 reduction catalysts. Nat Mater 17:301–307.
Wang C, Xie ZG, deKrafft KE, Lin WL (2011) Doping metal-organic frameworks for water oxidation, carbon dioxide reduction, and organic photocatalysis. J Am Chem Soc 133:13445–13454
Ryu UJ, Kim SJ, Lim HK, Kim H, Choi KM, Kang JK (2017) Synergistic interaction of Re complex and amine functionalized multiple ligands in metal-organic frameworks for conversion of carbon dioxide. Sci Rep 7:8
Choi KM, Kim D, Rungtaweevoranit B, Trickett CA, Barmanbek JTD, Alshammari AS, Yang PD, Yaghi OM (2017) Plasmon-enhanced photocatalytic CO2 conversion within metal organic frameworks under visible light. J Am Chem Soc 139:356–362.
Kornienko N, Zhao YB, Kiley CS, Zhu CH, Kim D, Lin S, Chang CJ, Yaghi OM, Yang PD (2015) Metal-organic frameworks for electrocatalytic reduction of carbon dioxide. J Am Chem Soc 137:14129–14135
Hod I, Sampson MD, Deria P, Kubiak CP, Farha OK, Hupp JT (2015) Fe-porphyrin-based metal-organic framework films as high-surface concentration, heterogeneous catalysts for electrochemical reduction of CO2. ACS Catal 5:6302–6309.
Jain SL, Sain B (2002) Ruthenium catalyzed oxidation of tertiary nitrogen compounds with molecular oxygen: an easy access to N-oxides under mild conditions. Chem Commun 10:1040–1041
Bass JS, Kevan L (1990) Electron-spin-resonance and electron-spin echo spectroscopic studies of paramagnetic rhodium species produced in RhCa-X zeolite during ethylene dimerization - evidence for a sigma-bonded intermediate. J Phys Chem 94:1483–1489
Sava Gallis DF, Parkes MV, Greathouse JA, Zhang XY, Nenoff TM (2015) Enhanced O2 selectivity versus N2 by partial metal substitution in Cu-BTC. Chem Mater 27:2018–2025.
Gul-E-Noor F, Jee B, Mendt M, Himsl D, Poppl A, Hartmann M, Haase J, Krautscheid H, Bertmer M (2012) Formation of mixed metal Cu3–XZnx(Btc)(2) frameworks with different zinc contents: incorporation of Zn2+ into the metal-organic framework structure as studied by solid-state NMR. J Phys Chem C 116:20866–20873.
Nickerl G, Stoeck U, Burkhardt U, Senkovska I, Kaskel S (2014) A catalytically active porous coordination polymer based on a dinuclear rhodium paddle-wheel unit. J Mater Chem A 2:144–148
Teo JM, Coghlan CJ, Evans JD, Tsivion E, Head-Gordon M, Sumby CJ, Doonan CJ (2016) Hetero-bimetallic metal-organic polyhedra. Chem Commun 52:276–279
Zhang WH, Chen ZH, Al-Naji M, Guo PH, Cwik S, Halbherr O, Wang YM, Muhler M, Wilde N, Glaser R, Fischer RA (2016) Simultaneous introduction of various palladium active sites into MOF via one-pot synthesis: Pd@Cu3–XPdx(BTC)(2)(N). Dalton Trans 45:14883–14887.
Xamena F, Abad A, Corma A, Garcia H (2007) MOFs as catalysts: activity, reusability and shape-selectivity of a Pd-containing MOF. J Catal 250:294–298
Schuster S, Klemm E, Bauer M (2012) The Role of Pd2+/Pd0 in hydrogenation by Pd(2-Pymo)(2)(N): an X-ray absorption and IR spectroscopic study. Chem-Eur J 18:15831–15837
Opelt S, Krug V, Sonntag J, Hunger M, Klemm E (2012) Investigations on stability and reusability of Pd(2-Pymo)(2)(N) as hydrogenation catalyst. Microporous Mesoporous Mater 147:327–333
Sabo M, Henschel A, Froede H, Klemm E, Kaskel S (2007) Solution infiltration of palladium into MOF-5: synthesis, physisorption and catalytic properties. J Mater Chem 17:3827–3832
Gole B, Sanyal U, Banerjee R, Mukherjee PS (2016) High loading of Pd nanoparticles by interior functionalization of MOFs for heterogeneous catalysis. Inorg Chem 55:2345–2354
Wang C, Zhang HY, Feng C, Gao ST, Shang NZ, Wang Z (2015) Multifunctional Pd@MOF core-shell nanocomposite as highly active catalyst for p-nitrophenol reduction. Catal Commun 72:29–32
Yuan BZ, Pan YY, Li YW, Yin BL, Jiang HF (2010) A highly active heterogeneous palladium catalyst for the Suzuki-Miyaura and Ullmann coupling reactions of aryl chlorides in aqueous media. Angew Chem-Int Ed 49:4054–4058
Oien S, Wragg D, Reinsch H, Svelle S, Bordiga S, Lamberti C, Lillerud KP (2014) Detailed structure analysis of atomic positions and defects in zirconium metal-organic frameworks. Cryst Growth Des 14:5370–5372
Shearer GC, Chavan S, Ethiraj J, Vitillo JG, Svelle S, Olsbye U, Lamberti C, Bordiga S, Lillerud KP (2014) Tuned to perfection: ironing out the defects in metal-organic framework UiO-66. Chem Mater 26:4068–4071
Shearer GC, Chavan S, Bordiga S, Svelle S, Olsbye U, Lillerud KP (2016) Defect engineering: tuning the porosity and composition of the metal-organic framework Uio-66 via modulated synthesis. Chem Mater 28:3749–3761
Shearer GC, Vitillo JG, Bordiga S, Svelle S, Olsbye U, Lillerud KP (2016) Functionalizing the defects: postsynthetic ligand exchange in the metal organic framework UiO-66. Chem Mater 28:7190–7193
Karagiaridi O, Vermeulen NA, Klet RC, Wang TC, Moghadam PZ, Al-Juaid SS, Stoddart JF, Hupp JT, Farha OK (2015) Functionalized defects through solvent-assisted linker exchange: synthesis, characterization, and partial postsynthesis elaboration of a metal-organic framework containing free carboxylic acid moieties. Inorg Chem 54:1785–1790
Brozek CK, Dinca M (2013) Ti3+-, V2+/3+-, Cr2+/3+-, Mn2+-, and Fe2+-substituted MOF-5 and redox reactivity in Cr- and Fe-MOF-5. J Am Chem Soc 135:12886–12891
Kim M, Cahill JF, Fei HH, Prather KA, Cohen SM (2012) Postsynthetic ligand and cation exchange in robust metal-organic frameworks. J Am Chem Soc 134:18082–18088
Gu ZG, Heinke L, Wöll C, Neumann T, Wenzel W, Li Q, Fink K, Gordan OD, Zahn DRT (2015) Experimental and theoretical investigations of the electronic band structure of metal-organic frameworks of HKUST-1 Type. Appl Phys Lett 107:5
Wang ZB, Sezen H, Liu JX, Yang CW, Roggenbuck SE, Peikert K, Froba M, Mavrantonakis A, Supronowicz B, Heine T, Gliemann H, Wöll C (2015) Tunable coordinative defects in UHM-3 surface-mounted MOFs for gas adsorption and separation: a combined experimental and theoretical study. Microporous Mesoporous Mater 207:53–60
Shekhah O, Swaidan R, Belmabkhout Y, du Plessis M, Jacobs T, Barbour LJ, Pinnau I, Eddaoudi M (2014) The liquid phase epitaxy approach for the successful construction of ultra-thin and defect-free ZIF-8 membranes: pure and mixed gas transport study. Chem Commun 50:2089–2092
Liu GP, Chernikova V, Liu Y, Zhang K, Belmabkhout Y, Shekhah O, Zhang C, Yi SL, Eddaoudi M, Koros WJ (2018) Mixed matrix formulations with MOF molecular sieving for key energy-intensive separations. Nat Mater 17:283–289
Wang ZB, Knebel A, Grosjean S, Wagner D, Brase S, Wöll C, Caro J, Heinke L (2016) Tunable molecular separation by nanoporous membranes. Nat Commun 7:7
Muller K, Knebel A, Zhao FL, Bleger D, Caro J, Heinke L (2017) Switching thin films of azobenzene-containing metal-organic frameworks with visible light. Chem-Eur J 23:5434–5438
Hurrle S, Friebe S, Wohlgemuth J, Wöll C, Caro J, Heinke L (2017) Sprayable, large-area metal-organic framework films and membranes of varying thickness. Chem-Eur J 23:2294–2298
Beyzavi MH, Vermeulen NA, Zhang KN, So M, Kung CW, Hupp JT, Farha OK (2016) Liquid-phase epitaxially grown metal-organic framework thin films for efficient tandem catalysis through site-isolation of catalytic centers. ChemPlusChem 81:708–713
Katz MJ, Moon SY, Mondloch JE, Beyzavi MH, Stephenson CJ, Hupp JT, Farha OK (2015) Exploiting parameter space in MOFs: A 20-fold enhancement of phosphate-ester hydrolysis with UiO-66-NH2. Chem Sci 6:2286–2291
Evans JD, Sumby CJ, Doonan CJ (2014) Post-synthetic metalation of metal-organic frameworks. Chem Soc Rev 43:5933–5951
Heinke L, Gu ZG, Wöll C (2014) The surface barrier phenomenon at the loading of metal-organic frameworks. Nat Commun 5:4562
Seo JS, Whang D, Lee H, Jun SI, Oh J, Jeon YJ, Kim K (2000) A homochiral metal-organic porous material for enantioselective separation and catalysis. Nature 404:982–986
Evans OR, Ngo HL, Lin WB (2001) Chiral porous solids based on lamellar lanthanide phosphonates. J Am Chem Soc 123:10395–10396
Wu CD, Hu A, Zhang L, Lin WB (2005) Homochiral porous metal-organic framework for highly enantioselective heterogeneous asymmetric catalysis. J Am Chem Soc 127:8940–8941
Cho SH, Ma BQ, Nguyen ST, Hupp JT, Albrecht-Schmitt TE (2006) A metal-organic framework material that functions as an enantioselective catalyst for olefin epoxidation. Chem Commun 24:2563–2565
Banerjee M, Das S, Yoon M, Choi HJ, Hyun MH, Park SM, Seo G, Kim K (2009) Postsynthetic modification switches an achiral framework to catalytically active homochiral metal-organic porous materials. J Am Chem Soc 131:7524–7525
Ma LQ, Falkowski JM, Abney C, Lin WB (2010) A series of isoreticular chiral metal-organic frameworks as a tunable platform for asymmetric catalysis. Nat Chem 2:838–846
Lun DJ, Waterhouse GIN, Telfer SG (2011) A general thermolabile protecting group strategy for organocatalytic metal-organic frameworks. J Am Chem Soc 133:5806–5809
Dang DB, Wu PY, He C, Xie Z, Duan CY (2010) Homochiral metal-organic frameworks for heterogeneous asymmetric catalysis. J Am Chem Soc 132:14321–14323
Ma LQ, Abney C, Lin WB (2009) Enantioselective catalysis with homochiral metal-organic frameworks. Chem Soc Rev 38:1248–1256
Dybtsev DN, Nuzhdin AL, Chun H, Bryliakov KP, Talsi EP, Fedin VP, Kim K (2006) A homochiral metal-organic material with permanent porosity, enantioselective sorption properties, and catalytic activity. Angew Chem-Int Ed 45:916–920
Li G, Yu WB, Ni J, Liu TF, Liu Y, Sheng EH, Cui Y (2008) Self-assembly of a homochiral nanoscale metallacycle from a metallosalen complex for enantioselective separation. Angew Chem-Int Ed 47:1245–1249
Nickerl G, Henschel A, Grunker R, Gedrich K, Kaskel S (2011) Chiral metal-organic frameworks and their application in asymmetric catalysis and stereoselective separation. Chem Ing Tech 83:90–103
Liu Y, Xuan WM, Cui Y (2010) Engineering homochiral metal-organic frameworks for heterogeneous asymmetric catalysis and enantioselective separation. Adv Mater 22:4112–4135
Gu ZG, Zhan CH, Zhang J, Bu XH (2016) Chiral chemistry of metal-camphorate frameworks. Chem Soc Rev 45:3122–3144
Liu B, Shekhah O, Arslan HK, Liu JX, Wöll C, Fischer RA (2012) Enantiopure metal-organic framework thin films: oriented SURMOF growth and enantioselective adsorption. Angew Chem-Int Ed 51:807–810
Gu ZG, Burck J, Bihlmeier A, Liu JX, Shekhah O, Weidler PG, Azucena C, Wang ZB, Heissler S, Gliemann H, Klopper W, Ulrich AS, Wöll C (2014) Oriented circular dichroism analysis of chiral surface-anchored metal-organic frameworks grown by liquid-phase epitaxy and upon loading with chiral guest compounds. Chem-Eur J 20:9879–9882
Gu ZG, Grosjean S, Brase S, Wöll C, Heinke L (2015) Enantioselective adsorption in homochiral metal-organic frameworks: the pore size influence. Chem Commun 51:8998–9001
Gu ZG, Fu H, Neumann T, Xu ZX, Fu WQ, Wenzel W, Zhang L, Zhang J, Wöll C (2016) Chiral porous metacrystals: employing liquid-phase epitaxy to assemble enantiopure metal-organic nanoclusters into molecular framework pores. ACS Nano 10:977–983
Hermes S, Schroter MK, Schmid R, Khodeir L, Muhler M, Tissler A, Fischer RW, Fischer RA (2005) Metal@Mof: loading of highly porous coordination polymers host lattices by metal organic chemical vapor deposition. Angew Chem-Int Ed 44:6237–6241
Esken D, Turner S, Lebedev OI, Van Tendeloo G, Fischer RA (2010) Au@ZIFs: stabilization and encapsulation of cavity-size matching gold clusters inside functionalized zeolite imidazolate frameworks, Zifs. Chem Mater 22:6393–6401
Muller M, Turner S, Lebedev OI, Wang YM, van Tendeloo G, Fischer RA (2011) Au@MOF-5 and Au/MOx@MOF-5 (M = Zn, Ti; X = 1, 2): preparation and microstructural characterisation. Eur J Inorg Chem 2011:1876–1887
Hermannsdorfer J, Friedrich M, Miyajima N, Albuquerque RQ, Kummel S, Kempe R (2012) Ni/Pd@Mil-101: synergistic catalysis with cavity-conform Ni/Pd nanoparticles. Angew Chem-Int Ed 51:11473–11477
Zhu QL, Li J, Xu Q (2013) Immobilizing metal nanoparticles to metal-organic frameworks with size and location control for optimizing catalytic performance. J Am Chem Soc 135:10210–10213
Huang YB, Zhang YH, Chen XX, Wu DS, Yi ZG, Cao R (2014) Bimetallic alloy nanocrystals encapsulated in ZIF-8 for synergistic catalysis of ethylene oxidative degradation. Chem Commun 50:10115–10117
Zhou JJ, Wang P, Wang CX, Goh YT, Fang Z, Messersmith PB, Duan HW (2015) Versatile core-shell nanoparticle@metal-organic framework nanohybrids: exploiting mussel-inspired polydopamine for tailored structural Integration. ACS Nano 9:6951–6960
Chen LY, Chen XD, Liu HL, Li YW (2015) Encapsulation of mono- or bimetal nanoparticles inside metal-organic frameworks via in situ incorporation of metal precursors. Small 11:2642–2648
Rosler C, Esken D, Wiktor C, Kobayashi H, Yamamoto T, Matsumura S, Kitagawa H, Fischer RA (2014) Encapsulation of bimetallic nanoparticles into a metal-organic framework: preparation and microstructure characterization of Pd/Au@ZIF-8. Eur J Inorg Chem 2014:5514–5521
Rosler C, Dissegna S, Rechac VL, Kauer M, Guo PH, Turner S, Ollegott K, Kobayashi H, Yamamoto T, Peeters D, Wang YM, Matsumura S, Van Tendeloo G, Kitagawa H, Muhler M, Xamena F, Fischer RA (2017) Encapsulation of bimetallic metal nanoparticles into robust zirconium-based metal-organic frameworks: evaluation of the catalytic potential for size-selective hydrogenation. Chem-Eur J 23:3583–3594
Zhang WN, Lu G, Cui CL, Liu YY, Li SZ, Yan WJ, Xing C, Chi YR, Yang YH, Huo FW (2014) A family of metal-organic frameworks exhibiting size-selective catalysis with encapsulated noble-metal nanoparticles. Adv Mater 26:4056–4060
Lu G, Li SZ, Guo Z, Farha OK, Hauser BG, Qi XY, Wang Y, Wang X, Han SY, Liu XG, DuChene JS, Zhang H, Zhang QC, Chen XD, Ma J, Loo SCJ, Wei WD, Yang YH, Hupp JT, Huo FW (2012) Imparting functionality to a metal-organic framework material by controlled nanoparticle encapsulation. Nat Chem 4:310–316
Zhan WW, Kuang Q, Zhou JZ, Kong XJ, Xie ZX, Zheng LS (2013) Semiconductor@metal-organic framework core-shell heterostructures: a case of ZnO@ZIF-8 nanorods with selective photoelectrochemical response. J Am Chem Soc 135:1926–1933
Li XY, Pi YH, Hou QQ, Yu H, Li Z, Li YW, Xiao J (2018) Amorphous TiO2@NH2-MIL-125(Ti) homologous MOF-encapsulated heterostructures with enhanced photocatalytic activity. Chem Commun 54:1917–1920
Esken D, Noei H, Wang YM, Wiktor C, Turner S, Van Tendeloo G, Fischer RA (2011) Zno@Zif-8: stabilization of quantum confined ZnO nanoparticles by a zinc methylimidazolate framework and their surface structural characterization probed by CO2 adsorption. J Mater Chem 21:5907–5915.
Guo W, Chen Z, Yang CW, Neumann T, Kuebel C, Wenzel W, Welle A, Pfleging W, Shekhah O, Wöll C, Redel E (2016) Bi2O3 nanoparticles encapsulated in surface mounted metal-organic framework thin films. Nanoscale 8:6468–6472
Domenech A, Garcia H, Domenech-Carbo MT, Xamena F (2007) Electrochemistry of metal-organic frameworks: a description from the voltammetry of microparticles approach. J Phys Chem C 111:13701–13711
Babu KF, Kulandainathan MA, Katsounaros I, Rassaei L, Burrows AD, Raithby PR, Marken F (2010) Electrocatalytic activity of basolite (Tm) F300 metal-organic-framework structures. Electrochem Commun 12:632–635
Dragasser A, Shekhah O, Zybaylo O, Shen C, Buck M, Wöll C, Schlettwein D (2012) Redox mediation enabled by immobilised centres in the pores of a metal-organic framework grown by liquid phase epitaxy. Chem Commun 48:663–665
Mugnaini V, Tsotsalas M, Bebensee F, Grosjean S, Shahnas A, Brase S, Lahann J, Buck M, Wöll C (2014) Electrochemical investigation of covalently post-synthetic modified surgel coatings. Chem Commun 50:11129–11131
Liu JX, Paradinas M, Heinke L, Buck M, Ocal C, Mugnaini V, Wöll C (2016) Film quality and electronic properties of a surface-anchored metal-organic framework revealed by using a multi-technique approach. ChemElectroChem 3:713–718
Ye L, Liu JX, Gao Y, Gong CH, Addicoat M, Heine T, Wöll C, Sun LC (2016) Highly oriented MOF thin film-based electrocatalytic device for the reduction of CO2 to CO exhibiting high faradaic efficiency. J Mater Chem A 4:15320–15326.
Zhang LP, Cui P, Yang HB, Chen JZ, Xiao FX, Guo YY, Liu Y, Zhang WN, Huo FW, Liu B (2016) Metal-organic frameworks as promising photosensitizers for photoelectrochemical water splitting. Adv Sci 3:6
Liu JX, Zhou WC, Liu JX, Howard I, Kilibarda G, Schlabach S, Coupry D, Addicoat M, Yoneda S, Tsutsui Y, Sakurai T, Seki S, Wang ZB, Lindemann P, Redel E, Heine T, Wöll C (2015) Photoinduced charge-carrier generation in epitaxial MOF thin films: high efficiency as a result of an indirect electronic band gap? Angew Chem-Int Ed 54:7441–7445
Acknowledgements
We acknowledge financial support from the German Research Foundation (DFG), as well as from the “Science and Technology of Nanosystems” Programme (Project No. 432202).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, Y., Wöll, C. Chemical Reactions at Isolated Single-Sites Inside Metal–Organic Frameworks. Catal Lett 148, 2201–2222 (2018). https://doi.org/10.1007/s10562-018-2432-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-018-2432-2