
Abstract
Treatment of malignant disease is of paramount importance in modern medicine. In 2012, it was estimated that 162,000 people died from cancer in the UK which illustrates a fundamental problem. Traditional treatments for cancer have various drawbacks, and this creates a considerable need for specific, molecular targets to overcome cancer spread. Epithelial protein lost in neoplasm (EPLIN) is an actin-associated molecule which has been implicated in the development and progression of various cancers including breast, prostate, oesophageal and lung where EPLIN expression is frequently lost as the cancer progresses. EPLIN is important in the regulation of actin dynamics and has multiple associations at epithelial cells junctions. Thus, EPLIN loss in cancer may have significant effects on cancer cell migration and invasion, increasing metastatic potential. Overexpression of EPLIN has proved to be an effective tool for manipulating cancerous traits such as reducing cell growth and cell motility and rendering cells less invasive illustrating the therapeutic potential of EPLIN. Here, we review the current state of knowledge of EPLIN, highlighting EPLIN involvement in regulating cytoskeletal dynamics, signalling pathways and implications in cancer and metastasis.
Similar content being viewed by others


1 Introduction
The incidence of cancer is slowly rising and has become a global burden. A fundamental reason why cancer is such a problem is because of its ability to spread, invade surrounding tissue and potentially form secondary cancers at distinct sites around the body by metastasis. Cancer hallmarks include uncontrolled cell growth and evasion of cell death, and this ultimately can lead to tumour formation. According to the World Health Organisation (WHO), 8.2 million people died from cancer in 2012 worldwide [1]. In the UK alone, mortality rates reached 162,000 annual deaths [2]. This illustrates a considerable need for better treatment, diagnosis and management of the disease. Epithelial protein lost in neoplasm (EPLIN) is a molecule involved in regulation of the actin cytoskeleton and has been implicated in the development and progression of various cancer types, displaying frequent downregulation or loss in cancer, creating a potential for prognostic targeting and as a tumour suppressor. This current review discusses EPLIN’s role in actin dynamics and in the pathophysiology of cancer development and progression.
2 Epithelial protein lost in neoplasm
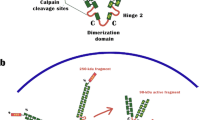
EPLIN is a cytoskeletal, actin-binding protein encoded by the LIMA1 gene. EPLIN was initially identified in oral cancers for its differential expression between normal oral epithelial cells and human papilloma virus (HPV)-immortalised oral epithelial cells [3]. EPLIN exists as two distinct isoforms, a 600 amino acid EPLINα isoform and a larger 759 amino acid EPLINβ isoform, generated from an alternative pre-mRNA splicing event (see Fig. 1) [4]. The EPLINα isoform has been implicated in the progression of various cancers, and this was initially recognised in oral cancer, breast, prostate and xenograft tumours where EPLIN expression was either downregulated or completely abolished [4]. The amino acid sequence of EPLIN is characterised by a single centrally located LIM domain which supposedly aids structural self-dimerisation and contains subdomains for zinc binding (see Fig. 2) [4, 5]. LIM-domain-containing proteins are frequently present in molecules responsible for cytoskeletal organisation, such as the focal adhesion phosphoprotein, paxillin [6]. EPLIN is important in the regulation of actin dynamics and aids actin filament bundle assembly, and the amino terminal of the EPLIN protein structure is essential for this localisation to the actin cytoskeleton [7]. The EPLIN genomic structure consists of 11 exons and ten introns, with exons 1–3 only present in EPLINβ and EPLINα utilising exons 4–11 of LIMA1 for transcription [5]. The EPLIN gene has two separate promoter regions; the EPLINβ promoter is near the start of the gene in exon 1, whilst EPLINα initiates ∼50 kb downstream near the end of intron 3, prior to exon 4 and at amino acid position 161 in the EPLINβ protein (see Fig. 1) [5]. Sequence analysis has revealed that EPLIN is conserved across species with EPLINα and EPLINβ isoforms present in mouse, displaying 77 and 75 % identity similarity for human EPLINα and EPLINβ, respectively (see Fig. 3) [8]. A role for EPLIN has also been suggested in muscle development in pigs, where EPLIN displayed a temporal expression pattern with only the EPLINα isoform present in developing skeletal muscle [9]. Since the discovery of EPLIN, our lab has shown that aberrant EPLIN expression is associated with the progression of various cancer types including breast, oesophageal, pulmonary and prostate cancer. EPLINα levels decrease as the cancer progresses and becomes more advanced, giving EPLINα potential to provide prognostic value, and overexpression analysis suggests that EPLINα is a putative tumour suppressor [10–14]. The described loss of EPLIN in cancer has functional implications on the actin cytoskeleton and may contribute to enhanced metastatic potential of cancer cells.

Schematic diagram of the LIMA1 genomic structure and EPLIN structural isoforms. The LIMA1 gene consists of 11 exons and ten introns. EPLINα differs from EPLINβ at the amino terminus where an additional 160 amino acids are present in EPLINβ. Shown below EPLINβ is the 52-amino acid centrally located LIM domain common to both EPLIN isoforms. Adapted from [4]

Protein structure of EPLIN LIM domain (PDB ID=2D8Y). Protein structure of the EPLIN centrally located LIM domain. Zinc-binding domains depicted. This domain may aid self-dimerisation. Image generated using UCSF Chimera software

ClustalW protein alignment of human, mouse and pig EPLINβ. Areas of amino acids that are conserved across species are highlighted. The region shown is amino side of the EPLINβ protein, where EPLINα originates at amino acid (AA) p. 161. ClustalW generated using BioEdit Biological Sequence Alignment software
2.1 The epithelial protein lost in neoplasm interactome: regulation in actin dynamics
EPLIN has a number of functional partners (see Fig. 4/Table 1), and the globular protein actin is central to the function of EPLIN. EPLIN has two functional actin-binding sites which flank the central LIM domain, and it is this binding capacity that engenders actin cross linking and actin filament bundle assembly [15]. A fibrillar pattern is displayed by both isoforms, and expression of EPLINα enhances the size and number of actin filament stress fibres and can also inhibit membrane ruffling via the signalling GTPase, Rac1 [15]. EPLIN therefore directly interacts with actin which suggests a possible role for EPLIN in cell migration, adhesion and cell morphology. Actin is an abundant, multifunctional protein responsible for cell migration in eukaryotic cells. Actin is part of the cytoskeletal network which consists of microtubules, microfilaments and intermediate filaments which are vital for cellular functions. Actin exists as monomers (G-actin) and filamentous polymers (F-actin) and is important for physiological functions including cell locomotion, cytokinesis, maintenance of cell shape and muscle contraction [24]. Transcription of the LIMA1 gene is suggested to be primarily controlled by monomeric G actin, with the actin–MAL–SRF signalling pathway regulating EPLIN production [25]. Maul et al. [15] illustrated that EPLINα has three significant features: EPLINα has at least one binding site for actin and can cross link and bundle actin filaments, EPLINα stabilises actin filaments in vitro, and EPLINα inhibits branching nucleation of actin filaments by the Arp 2/3 complex [15]. Therefore, this suggests that EPLIN may orchestrate actin filament dynamics by stabilising actin cytoskeletal networks [15]. Additionally, EPLIN has been shown to form part of an actin-remodelling complex composing of EPLIN, β-actin, γ-actin and gelsolin which co-localises at the plasma membrane to the tumour suppressor, phosphatase and tensin homolog (PTEN) [26]. PTEN is a well-established tumour suppressor molecule, so this asks the question of whether the interaction of PTEN and the actin-remodelling complex is itself an element suppressing the development of neoplastic tissue and whether any interruption of these complexes may promote cancer progression. Based on these findings, EPLIN accommodates actin to accomplish various actin-related cellular processes including cell motility and migration and cell junctional adhesion [17]. There is increasing evidence to suggest that EPLIN regulates actin structures in cooperation with the signal transduction adaptor protein, paxillin. When EPLIN is overexpressed, paxillin exhibits an increased staining pattern for both human endothelial cells line (HECV) and PC-3 cells [12, 13]. This co-localisation pattern is also observed in cultured human mesangial cells at focal adhesion sites, and co-immunoprecipitation results confirm an association between the two molecules [16]. EPLIN and paxillin may form a complex and potentially stabilise focal adhesions to co-ordinate actin dynamics in a complimentary manner. Given EPLIN’s role in actin dynamics, it is strongly implicated in cellular processes including cell migration and invasion, and thus, downregulation or loss of EPLIN expression in cancer may likely affect the metastatic potential of cancer cells. Actin and EPLIN are located in epithelial cells at the adherens junction (AJ) and contribute to functional cellular adhesion between adjacent cells.

EPLIN predicted functional partners. EPLIN (LIMA1) has various associations including cadherin and catenin molecules which contribute to cytoskeleton regulation. LIMA1 LIM domain and actin binding 1; CDH1 cadherin 1; CTNNA1 catenin-α1; CDH1 cadherin 1; CTNND1 catenin-δ1; CTNNB1 catenin-β1; UBC ubiquitin C; PTPLAD1 protein tyrosine phosphatase-like A domain-containing 1; ARPC1A actin-related protein 2/3 complex, subunit 1A; ATP6V1B1 ATPase, H+ transporting, lysosomal 56–58 kDa, V1 subunit B1; YWHAH tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein; SVIL supervillin. Image generated and extracted from online STRING database
2.2 The adherens junction
The AJ is a type of anchoring junction found predominantly in epithelial cells, also referred to as the zonula adherens, which functionally link the actin cytoskeleton together in cells via linker molecules. EPLIN is an actin-binding protein, which functions to bundle actin filaments; therefore, EPLIN presence is required at AJ along with filamentous actin. AJ contains various protein complexes along with EPLIN, and these include cadherins, catenins and p120 catenins (see Fig. 5) [28]. Within the AJ, cadherins and catenins associate together to form the cadherin–catenin complex and EPLIN provides a direct physical link for this complex to the actin cytoskeleton [17]. The cadherin–catenin complex is composed of E-cadherin, β-catenin and α-catenin, with E-cadherin positioned between adjacent cells and the catenins positioned in the cytoplasmic space of each cell [29]. E-cadherin binds directly to β-catenin which sequentially binds α-catenin, generating the cadherin–catenin complex (see Fig. 5) [30]. α-Catenin is a crucial player at the AJ and was initially recognised as responsible for providing the bridge between the cadherin–catenin complex and actin [28, 31]. This principle, however, has come under scrutiny, as direct in vitro binding between α-catenin and actin has never been detected [32, 33]. Cavey and co-workers [34] realised that α-catenin is not essential for E-cadherin stability, as complexes of α-catenin and E-cadherin were detected in RNAi α-catenin embryos [31]. Therefore, it is apparent that cadherin–actin interaction is regulated not only by α-catenin but by a number of actin-binding proteins that are associated with α-catenin, including EPLIN [30, 35]. Abe and Takeichi [17] demonstrated, by immunoprecipitation and GST pull-down assays, that both EPLIN isoforms directly interact with the α-catenin VH3 plus C-terminal region to generate a cadherin–catenin–EPLIN–actin complex at cell junctions [17]. When EPLIN is depleted, the bridge to F-actin was unable to form due to loss of organisation of the apical actin belt, with punctate accumulation of E-cadherin at cell junctional points [17]. This illustrates the importance of EPLIN in producing functional epithelial junctions. Additional molecules involved in regulating the AJ include the membrane cytoskeletal protein, vinculin, and the actin filament-binding protein, afadin [36, 37]. Recent work from Taguchi et al. [35] illustrated that EPLIN and vinculin may collaborate together in AJ formation via binding α-catenin either together or individually and cooperatively aid junctional adhesion [35]. The cooperation of EPLIN and vinculin in cellular adhesion is also evident in endothelial cells. At the endothelial AJ, the endothelial E-cadherin homolog, VE-cadherin, interacts with β- and γ-catenins, which sequentially bind α-catenin and EPLIN in an analogous fashion to epithelial cells [38]. This allows the recruitment of vinculin and ultimately promotes strengthening of inter-endothelial junctions [38]. The authors propose a role for EPLIN in tension dissemination at the endothelial AJ in a mechanosensory mechanism [38]. The machinery from actomyosin exerts tension through EPLIN, which causes α-catenin to adopt a more accessible conformation, revealing a vinculin-binding site and allowing vinculin recruitment and actin association at endothelial cell–cell AJ [39]. This mechanotransduction mechanism consisting of EPLIN and α-catenin suggests that the endothelial AJ is regulated in a spatial and temporal fashion [39]. Finally, EPLIN also appears to be important for attachment to F-actin in endothelial cells; when EPLIN expression is downregulated, the organisation of F-actin is considerably disrupted, leading to multiple holes in the actin cytoskeleton [38]. Altogether, this suggests that the AJs of epithelial and endothelial cells are orchestrated by various actin-binding, α-catenin-associated molecules and are dynamically regulated, with EPLIN having a critical role in cell adhesion, creating further implications of EPLIN loss in cancer.

Schematic representation of adherens junctions. The AJ between epithelial cells consists of various protein complexes to orchestrate actin cytoskeletal dynamics. The cadherin–catenin complex is associated with filamentous actin via EPLIN and/or vinculin which binds α-catenin in the cytoplasm. Adapted from [27]
2.3 Epithelial protein lost in neoplasm—a key player in cell division?
Cell division is the splitting of one cell into two, where biological information is passed onto daughter cells. For this process to successfully occur, various proteins need to functionally regulate the division and these include Rho GTPases, cyclin-dependant kinases, integrins, cdc42, focal adhesion kinases, myosin and the globular protein actin [40]. With this in mind, an actin-binding protein like EPLIN may potentially have a regulatory role in cell division. This has been recently shown using HeLa cells where EPLIN depletion resulted in large numbers of multinucleated cells, signifying cytokinesis failure during cell division [41]. In successful mitotic division, actin and myosin II accumulate at the cleavage furrow during cytokinesis and EPLIN loss compromised each protein’s ability to efficiently do this [41]. EPLIN appears to be important for the accumulation of other mitotic regulatory proteins including the GTPases RhoA and cdc42, where EPLIN depletion resulted in either a significantly reduced concentration of RhoA or a misplaced location of cdc42 at the cleavage furrow [41]. EPLIN aids this successful cell division in conjunction with a number of regulatory proteins including supervillin and the oncogenic kinesin, KIF14, suggesting a complex network of regulatory proteins at the cleavage furrow [18]. Altogether, this suggests that EPLIN may have an integral role in cytokinesis and loss may lead to aneuploidy and genomic instability of daughter cells [41]. Therefore, EPLIN is crucial to co-ordinate actin and myosin dynamics throughout cell division and loss in cancer cells could have downstream effects on successful cytokinesis, increasing their tendency to form a cancer [41].
2.4 Post-translational modification
Extracellular signal-regulated kinase (ERK) is a member of the mitogen-activated protein kinase (MAPK) family and is important in the regulation of actin organisation by phosphorylating various proteins including paxillin, focal adhesion kinase (FAK) and other protein kinases and nuclear transcription factors to co-ordinate cellular processes [42, 43]. ERK is implicated in cellular events including cell migration and may facilitate this by phosphorylation of actin-bundling proteins like EPLIN [20]. The protein structure of EPLIN has multiple putative phosphorylation sites (see Fig. 6), and it has been shown that ERK phosphorylates EPLIN at Ser360, Ser602 and Ser692 in vitro and in vivo [20]. Phosphorylation at the carboxy terminal of EPLIN decreases affinity to F-actin and thus provokes a reorganisation of the actin cytoskeleton, enhancing cell migration [20]. This implicates ERK in actin organisation and cell motility with EPLIN being a critical substrate for phosphorylation [20]. A recent study by Zhang et al. [44] illustrated that this ERK-mediated phosphorylation of EPLIN is itself regulated by epidermal growth factor (EGF) and revealed how targeting this signalling cascade can be manipulated to reduce epithelial–mesenchymal transition (EMT) and, thus, prostate cancer invasiveness [44]. ERK also plays a role in targeting EPLIN to focal adhesions and effects the interaction with paxillin; activation of the MEK–ERK pathway both reduced localisation of EPLIN to sites of focal adhesions and abolished paxillin interaction [16]. These data suggest that ERK is functionally related to EPLIN and provides a critical regulatory role for appropriate actin dynamics and may have implications in cancer progression.

Predicted phosphorylation sites in EPLINα. The protein structure of human EPLIN has multiple putative phosphorylation sites at all regions of the protein, including various sites where serine kinases would likely act. Predicted threonine and tyrosine phosphorylation sites not shown. The phosphorylated residue suggested in [20] is indicated. Phosphorylation status predicted using NetPhos 2.0 software
3 The role of epithelial protein lost in neoplasm in cancer
Cancer progression involves various cellular, morphological and molecular alterations which result in a transformed cellular phenotype, ultimately having the potential to invade surrounding tissue and disseminate throughout the body. Cancer treatment options remain largely unspecific and create various undesired side effects. Therefore, elucidating a molecular target for treating cancer, in addition to understanding the mechanism of cancer development, is crucial. EPLIN first received attention for its involvement in cancer in 1999 where EPLIN downregulation was described in various cancer cell lines [4]. Altogether, low levels of EPLIN transcript were found in 8/8 oral cancer cell lines, 5/6 breast cancer cell lines and 4/4 prostate cancer cell lines [4]. Using PC-3 and DU-145 prostate cancer cell lines, EPLIN expression was significantly reduced compared to primary prostate epithelial cells (PrEC), whereas the prostate specific antigen (PSA) positive LNCaP and LAPC4 prostate cancer cell lines failed to express EPLINα at all [4]. This notion of EPLIN loss is also seen in breast cancer where EPLIN expression in cell lines BT-20, SKBr-3, MCF-7, T-47D and MDA-MB-231 was either reduced or completely lost [4]. Lastly, the authors demonstrated EPLIN as a putative tumour suppressor molecule, where overexpression of EPLINα caused a reduction in cancer cell growth [4]. Interestingly, when EPLINα was depleted in breast cancer cell lines, EPLINβ either remained consistent or actually increased [4]. This illustrates the potential cancer protective effects that the EPLINα isoform may exert in various cancer cell systems. EPLIN overexpression has also proved effective in altering the growth phenotype and morphology in additional cell systems including anchorage-independent NIH3T3 transformed cells [7]. Using a soft agar assay and utilising the activated Cdc42 or the chimeric nuclear oncogene EWS/Fli-1 to transform NIH3T3 cells, EPLIN overexpression resulted in a ∼80 % decrease in colony formation for Cdc42 transformed cells, with a similar growth decrease in EWS/Fli-1 transformed cells [7]. Interestingly, EPLIN displayed heterogeneous staining throughout Ras cells rather than localisation to the actin cytoskeleton [7]. This implies that oncogenic transformation affects the EPLIN/actin architecture, and thus, the localisation of EPLIN to the actin cytoskeleton may be important to exert its suppressive ability [7].
3.1 Prostate cancer
There is increasing evidence to suggest that EPLIN is implicated in the development of prostate cancer and the process of EMT. EMT is a process where polarised epithelial cells are downregulated and subjected to biochemical and morphological changes. Epithelial cells can become transformed to a mesenchymal cell phenotype, losing their cell polarity and cell adhesion at cellular junctions [45]. The converted mesenchymal cell phenotype has a reorganised cytoskeleton and experiences alterations in cell signalling which engenders enhanced migratory and invasiveness capabilities and increased resistance to apoptosis [45, 46]. During these cellular changes, the actin molecular architecture is disrupted and protein complexes like the cadherin–catenin complex and epithelial markers are lost [47]. EPLIN is associated with the cadherin–catenin complex and contributes to functional cytoskeletal dynamics [17]. Zhang and co-workers [47] used biochemical and functional approaches to demonstrate that EPLIN is a negative regulator of EMT and invasiveness in prostate cancer (PCa) cells. EPLIN was significantly decreased in cells of more mesenchymal morphology (known as the androgen refractory cancer of the prostate (ARCaPM) cell lineage model), suggesting that EPLIN downregulation is directly implicated in EMT, along with the cadherin–catenin complex [47]. Depletion of EPLIN also provokes various other morphological changes including disassembly of AJ, increased migratory and invasive potential of cells in vitro, activation of β-catenin signalling, increased expression of vimentin, increased chemoresistance and decreased expression and nuclear translocation of E-cadherin [47]. Lastly, the authors used immunohistochemistry to show that EPLIN downregulation is correlated with cancer progression in multiple cancer models including lymph node metastasis in PCa, where EPLIN expression was significantly reduced [47]. Altogether, this illustrates that EPLIN may be involved in the regulation of EMT and PCa progression and loss of EPLIN can lead to diverse downstream cellular effects. EPLIN in PCa has also been recently evaluated by our laboratory using the classical PCa cell line, PC-3. By immunohistochemistry (IHC), EPLIN displayed a significant decrease in staining for tumour cells in comparison to normal (see Fig. 7a) [12]. This is accompanied by quantification of staining intensity within the cohort, where lower levels of EPLIN staining are associated with cancerous and higher-tumour-grade samples (see Fig. 7c, d) [12]. Overexpression analysis of EPLINα resulted in decreased growth rate in tumour cells in vitro, along with reduced invasiveness and cell adhesion to the extracellular matrix (ECM) [12]. Mice injected with PC-3 cells overexpressing EPLINα developed tumours at a markedly decreased rate in comparison to control [12]. Furthermore, cells overexpressing EPLINα displayed a greater staining pattern for the focal adhesion targeting protein, paxillin, implying that EPLIN may also be present at these plaques [4, 12]. Altogether, these results demonstrate the potential of EPLIN for monitoring PCa progression and how it can be manipulated to suppress PCa via impeding cancerous traits.

EPLIN profile in clinical prostate and breast cancer. Immunohistochemical staining (×20 objective magnification) of normal and cancerous a prostate and b breast clinical samples demonstrating EPLIN localisation and expressional differences. c, d Semi-quantitative analysis of EPLIN staining within prostate clinical cohort demonstrates that lower levels of EPLIN staining are associated with cancerous and higher-grade samples. e Within a clinical breast cancer cohort, lower transcript expression of EPLIN is seen in tumour samples compared to normal breast tissue and was associated with a higher grade (f), a poorer patient prognosis (g) and reduced overall survival rates (h). Figure modified from [10, 12]
3.2 Breast cancer
Our lab has recently evaluated EPLIN involvement in cancer progression in a number of model systems [10–14]. By comparing EPLINα IHC staining in normal vs tumour cells in breast cancer progression, EPLINα was found to be substantially weaker in tumour cells than in normal epithelial cells (see Fig. 7b) [10]. This correlated with lower EPLINα transcript in tumour samples compared to normal samples (see Fig. 7e) with lower EPLIN levels being associated with higher tumour grade, a poorer patient prognosis and reduced overall survival rates (see Fig. 7f–h) [10]. IHC analyses in breast cancer from additional research groups also show EPLIN loss as the tumour becomes more aggressive, specifically comparing EPLIN immunointensity of primary tumours vs tumours with lymph node metastases [47]. Lastly, in vitro and in vivo overexpression analysis of EPLIN highlighted significant reductions in cell growth and cell invasion using transfected breast cancer cell lines, and also, highly significant reductions in tumour size were observed in nude mice inoculated with EPLIN-α-transfected vs control MDA-MB-231 breast cancer cells [10].
3.3 Further pathological implications
Clinical implications for EPLIN also include oesophageal and pulmonary cancer (Table 2). Q-PCR analysis displayed reduced expression of EPLINα in an oesophageal cancer cohort for both cancerous tissue and cancer cell models [11]. With regard to tumour histological grade, tumour–node–metastasis (TNM) status, nodal status and survival status, EPLINα transcript generally decreased with levels significantly lower in patients who ultimately died from the cancer, suggesting that EPLINα is implicated in oesophageal cancer progression and may give an indication for cancer prognosis [11]. Overexpression analysis of EPLINα in the KYSE150 cell line resulted in decreased growth and invasiveness compared to normal, suggesting that EPLINα has tumour-suppressive ability by regulating cellular aggressiveness in oesophageal cancer [11]. In a pulmonary cancer cohort also conducted by our lab, Q-PCR analysis showed a reduction of EPLINα expression in tumour vs normal samples, where EPLIN was also reduced in later TNM stages and cancers with lymph node involvement [14]. Using the SKMES-1 cell line, overexpression of EPLINα via transfection in the pEF6 expression vector inhibited cell growth and cell motility [14]. In addition to the apparent molecular loss of EPLIN in various cancers, EPLIN also appears to be reduced at the protein level for colorectal cancer and squamous cell carcinoma of the head and neck (SCCHN), where IHC analysis revealed that EPLIN is decreased in cancers with lymph node metastases vs primary tumours [47].
Collectively, these studies suggest EPLIN may be a clinical indicator for cancer progression in addition to providing further evidence of a tumour-suppressive role for EPLINα in the regulation of cancer progression.
Finally, in addition to the implication of EPLIN in the spread and progression of cancer, a recent publication provides a link between EPLIN and renal diseases where patients with either membranoproliferative glomerulonephritis (MPGN) or IgA nephropathy had a decreased expression profile for EPLIN via IHC analysis [16]. This advocates the idea that EPLIN may be involved in the pathology of various disease states.
3.4 Angiogenesis
Angiogenesis is the formation of new blood vessels from pre-existing vessels and is essential for wound healing and normal growth and development. The angiogenic process is frequently utilised by cancer cells, by a means of metastasis, to reach secondary sites around the body and develop secondary tumours. Angiogenesis is therefore a critical factor when targeting cancer therapies. EPLINα demonstrates a suppressive role in angiogenesis, where overexpression analysis in the HECV endothelial cell line resulted in a reduced capacity to generate tubular structures in a Matrigel tubule formation assay when compared to vector controls [13]. This regulatory effect was also apparent in vivo where mice injected with HECV cells overexpressing EPLINα in conjunction with cancer cells developed tumours significantly slower than controls [13]. Forced expression also appears to exert an effect on cell matrix adhesion and migration capabilities in this cell line where cells overexpressing EPLINα both migrated at a significantly slower rate and were significantly less able to adhere to the Matrigel basement membrane [13]. This suppressive role in angiogenesis illustrates that EPLINα has potentially various regulatory mechanisms for reducing cancer metastasis and could be an effective target for cancer therapy.
4 Conclusions and outlook
The interaction of EPLIN and actin has provided an excellent model for investigating multiple aspects of cancer progression over the last decade. The discovery of EPLIN led to a subtle paradigm shift in structural view and organisation of cytoskeletal dynamics, with the acknowledgment that various actin-related molecules contribute to multiple dynamic processes underlying cellular migration and invasion. There is an established link between EPLIN and cancer progression with frequent downregulation of EPLIN in more aggressive cell lines, reduced staining in cancerous tissue samples and reduced growth potentials when there is forced expression of the EPLINα isoform in vitro and in vivo. EPLIN is functionally linked to molecules like actin and paxillin and has been implicated in a number of potential pathways to enhance metastatic potential (outlined in Fig. 8). However, the precise mechanistic action of EPLIN and, subsequently, how EPLIN loss contributes to the development of cancer remain elusive. Mechanistic investigations will therefore be crucial to elucidate the full importance of EPLIN in cancer pathophysiology.

Proposed EPLIN signalling pathways and implications for loss in cancer. When cancer is not present, EPLIN associates with the actin cytoskeleton linking the cadherin–catenin complex to F-actin via interaction with α-catenin. The signal transduction protein, paxillin, interacts with EPLIN in the cytoplasm, and this complex likely stabilises actin dynamics. ERK phosphorylates EPLIN regulating cell motility and migration. When cancer is present and EPLIN is lost, the actin cytoskeleton becomes less organised and this induces membrane ruffling. Paxillin targeting is likely lost reducing focal adhesion between the cadherin–catenin complex and actin. These molecular, cellular and morphological consequences may result in increased metastatic potential including enhanced cell migration and motility. Signalling pathways summarised from [12, 16, 20, 47]. Image generated using Pathway Builder 2.0 software
References
Stewart, B., & Wild, C. (2014). World Cancer Report 2014.
CancerResearchUK (2015). Cancer stats: cancer statistics for the UK. http://www.cancerresearchuk.org/cancer-info/cancerstats/. Accessed 20th November 2014.
Chang, D. D., Park, N. H., Denny, C. T., Nelson, S. F., & Pe, M. (1998). Characterization of transformation related genes in oral cancer cells. Oncogene, 16(15), 1921–1930. doi:10.1038/sj.onc.1201715.
Maul, R. S., & Chang, D. D. (1999). EPLIN, epithelial protein lost in neoplasm. Oncogene, 18(54), 7838–7841. doi:10.1038/sj.onc.1203206.
Chen, S., Maul, R. S., Kim, H. R., & Chang, D. D. (2000). Characterization of the human EPLIN (Epithelial Protein Lost in Neoplasm) gene reveals distinct promoters for the two EPLIN isoforms. Gene, 248(1–2), 69–76.
Brown, M. C., Perrotta, J. A., & Turner, C. E. (1996). Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. Journal of Cell Biology, 135(4), 1109–1123.
Song, Y., Maul, R. S., Gerbin, C. S., & Chang, D. D. (2002). Inhibition of anchorage-independent growth of transformed NIH3T3 cells by epithelial protein lost in neoplasm (EPLIN) requires localization of EPLIN to actin cytoskeleton. Molecular Biology of the Cell, 13(4), 1408–1416. doi:10.1091/mbc.01-08-0414.
Maul, R. S., Sachi Gerbin, C., & Chang, D. D. (2001). Characterization of mouse epithelial protein lost in neoplasm (EPLIN) and comparison of mammalian and zebrafish EPLIN. Gene, 262(1–2), 155–160.
Wang, H., Wang, H., Zhu, Z., Yang, S., Feng, S., & Li, K. (2007). Characterization of porcine EPLIN gene revealed distinct expression patterns for the two isoforms. Animal Biotechnology, 18(2), 101–108. doi:10.1080/10495390600864660.
Jiang, W. G., Martin, T. A., Lewis-Russell, J. M., Douglas-Jones, A., Ye, L., & Mansel, R. E. (2008). Eplin-alpha expression in human breast cancer, the impact on cellular migration and clinical outcome. Molecular Cancer, 7, 71. doi:10.1186/1476-4598-7-71.
Liu, Y., Sanders, A. J., Zhang, L., & Jiang, W. G. (2012). EPLIN-alpha expression in human oesophageal cancer and its impact on cellular aggressiveness and clinical outcome. Anticancer Research, 32(4), 1283–1289.
Sanders, A. J., Martin, T. A., Ye, L., Mason, M. D., & Jiang, W. G. (2011). EPLIN is a negative regulator of prostate cancer growth and invasion. Journal of Urology, 186(1), 295–301. doi:10.1016/j.juro.2011.03.038.
Sanders, A. J., Ye, L., Mason, M. D., & Jiang, W. G. (2010). The impact of EPLINalpha (Epithelial protein lost in neoplasm) on endothelial cells, angiogenesis and tumorigenesis. Angiogenesis, 13(4), 317–326. doi:10.1007/s10456-010-9188-7.
Liu, Y., Sanders, A. J., Zhang, L., & Jiang, W. G. (2012). Expression profile of Epithelial Protein Lost in Neoplasm-Alpha (EPLIN-α) in human pulmonary cancer and its impact on SKMES-1 cells in vitro. Journal of Cancer Therapy, 3, 452–459. doi:10.4236/jct.2012.324058.
Maul, R. S., Song, Y., Amann, K. J., Gerbin, S. C., Pollard, T. D., & Chang, D. D. (2003). EPLIN regulates actin dynamics by cross-linking and stabilizing filaments. Journal of Cell Biology, 160(3), 399–407. doi:10.1083/jcb.200212057.
Tsurumi, H., Harita, Y., Kurihara, H., Kosako, H., Hayashi, K., Matsunaga, A., et al. (2014). Epithelial protein lost in neoplasm modulates platelet-derived growth factor-mediated adhesion and motility of mesangial cells. Kidney International, 86(3), 548–557. doi:10.1038/ki.2014.85.
Abe, K., & Takeichi, M. (2008). EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proceedings of the National Academy of Sciences of the United States of America, 105(1), 13–19. doi:10.1073/pnas.0710504105.
Smith, T. C., Fang, Z., & Luna, E. J. (2010). Novel interactors and a role for supervillin in early cytokinesis. Cytoskeleton (Hoboken), 67(6), 346–364. doi:10.1002/cm.20449.
Karakose, E., Geiger, T., Flynn, K., Lorenz-Baath, K., Zent, R., Mann, M., et al. (2015). The focal adhesion protein PINCH-1 associates with EPLIN at integrin adhesion sites. Journal of Cell Science, 128(5), 1023–1033. doi:10.1242/jcs.162545.
Han, M. Y., Kosako, H., Watanabe, T., & Hattori, S. (2007). Extracellular signal-regulated kinase/mitogen-activated protein kinase regulates actin organization and cell motility by phosphorylating the actin cross-linking protein EPLIN. Molecular and Cellular Biology, 27(23), 8190–8204. doi:10.1128/mcb.00661-07.
Steder, M., Alla, V., Meier, C., Spitschak, A., Pahnke, J., Furst, K., et al. (2013). DNp73 exerts function in metastasis initiation by disconnecting the inhibitory role of EPLIN on IGF1R-AKT/STAT3 signaling. Cancer Cell, 24(4), 512–527. doi:10.1016/j.ccr.2013.08.023.
Seong, B. K., Lau, J., Adderley, T., Kee, L., Chaukos, D., Pienkowska, M., et al. (2014). SATB2 enhances migration and invasion in osteosarcoma by regulating genes involved in cytoskeletal organization. Oncogene. doi:10.1038/onc.2014.289.
Ohoka, A., Kajita, M., Ikenouchi, J., Yako, Y., Kitamoto, S., Kon, S., et al. (2015). EPLIN is a crucial regulator for extrusion of RasV12-transformed cells. Journal of Cell Science, 128(4), 781–789. doi:10.1242/jcs.163113.
Stricker, J., Falzone, T., & Gardel, M. L. (2010). Mechanics of the F-actin cytoskeleton. Journal of Biomechanics, 43(1), 9–14. doi:10.1016/j.jbiomech.2009.09.003.
Leitner, L., Shaposhnikov, D., Descot, A., Hoffmann, R., & Posern, G. (2010). Epithelial Protein Lost in Neoplasm alpha (Eplin-alpha) is transcriptionally regulated by G-actin and MAL/MRTF coactivators. Molecular Cancer, 9, 60. doi:10.1186/1476-4598-9-60.
Kim, J. S., Xu, X., Li, H., Solomon, D., Lane, W. S., Jin, T., et al. (2011). Mechanistic analysis of a DNA damage-induced, PTEN-dependent size checkpoint in human cells. Molecular and Cellular Biology, 31(13), 2756–2771. doi:10.1128/mcb.01323-10.
Ratheesh, A., & Yap, A. S. (2012). A bigger picture: classical cadherins and the dynamic actin cytoskeleton. Nature Reviews Molecular Cell Biology, 13(10), 673–679. doi:10.1038/nrm3431.
Meng, W., & Takeichi, M. (2009). Adherens junction: molecular architecture and regulation. Cold Spring Harbor Perspectives in Biology, 1(6), a002899. doi:10.1101/cshperspect.a002899.
Mege, R. M., Gavard, J., & Lambert, M. (2006). Regulation of cell-cell junctions by the cytoskeleton. Current Opinion in Cell Biology, 18(5), 541–548. doi:10.1016/j.ceb.2006.08.004.
Yonemura, S. (2011). Cadherin-actin interactions at adherens junctions. Current Opinion in Cell Biology, 23(5), 515–522. doi:10.1016/j.ceb.2011.07.001.
Nelson, W. J. (2008). Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochemical Society Transactions, 36(Pt 2), 149–155. doi:10.1042/bst0360149.
Drees, F., Pokutta, S., Yamada, S., Nelson, W. J., & Weis, W. I. (2005). Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell, 123(5), 903–915. doi:10.1016/j.cell.2005.09.021.
Yamada, S., Pokutta, S., Drees, F., Weis, W. I., & Nelson, W. J. (2005). Deconstructing the cadherin-catenin-actin complex. Cell, 123(5), 889–901. doi:10.1016/j.cell.2005.09.020.
Cavey, M., Rauzi, M., Lenne, P. F., & Lecuit, T. (2008). A two-tiered mechanism for stabilization and immobilization of E-cadherin. Nature, 453(7196), 751–756. doi:10.1038/nature06953.
Taguchi, K., Ishiuchi, T., & Takeichi, M. (2011). Mechanosensitive EPLIN-dependent remodeling of adherens junctions regulates epithelial reshaping. Journal of Cell Biology, 194(4), 643–656. doi:10.1083/jcb.201104124.
Geiger, B., Tokuyasu, K. T., Dutton, A. H., & Singer, S. J. (1980). Vinculin, an intracellular protein localized at specialized sites where microfilament bundles terminate at cell membranes. Proceedings of the National Academy of Sciences of the United States of America, 77(7), 4127–4131.
Sawyer, J. K., Harris, N. J., Slep, K. C., Gaul, U., & Peifer, M. (2009). The Drosophila afadin homologue Canoe regulates linkage of the actin cytoskeleton to adherens junctions during apical constriction. Journal of Cell Biology, 186(1), 57–73. doi:10.1083/jcb.200904001.
Chervin-Petinot, A., Courcon, M., Almagro, S., Nicolas, A., Grichine, A., Grunwald, D., et al. (2012). Epithelial protein lost in neoplasm (EPLIN) interacts with alpha-catenin and actin filaments in endothelial cells and stabilizes vascular capillary network in vitro. Journal of Biological Chemistry, 287(10), 7556–7572. doi:10.1074/jbc.M111.328682.
Gulino-Debrac, D. (2013). Mechanotransduction at the basis of endothelial barrier function. Tissue Barriers, 1(2), e24180. doi:10.4161/tisb.24180.
Heng, Y. W., & Koh, C. G. (2010). Actin cytoskeleton dynamics and the cell division cycle. International Journal of Biochemistry and Cell Biology, 42(10), 1622–1633. doi:10.1016/j.biocel.2010.04.007.
Chircop, M., Oakes, V., Graham, M. E., Ma, M. P., Smith, C. M., Robinson, P. J., et al. (2009). The actin-binding and bundling protein, EPLIN, is required for cytokinesis. Cell Cycle, 8(5), 757–764.
Pribic, J., & Brazill, D. (2012). Paxillin phosphorylation and complexing with Erk and FAK are regulated by PLD activity in MDA-MB-231 cells. Cellular Signalling, 24(8), 1531–1540. doi:10.1016/j.cellsig.2012.03.015.
Pearson, L. L., Castle, B. E., & Kehry, M. R. (2001). CD40-mediated signaling in monocytic cells: up-regulation of tumor necrosis factor receptor-associated factor mRNAs and activation of mitogen-activated protein kinase signaling pathways. International Immunology, 13(3), 273–283.
Zhang, S., Wang, X., Iqbal, S., Wang, Y., Osunkoya, A. O., Chen, Z., et al. (2013). Epidermal growth factor promotes protein degradation of epithelial protein lost in neoplasm (EPLIN), a putative metastasis suppressor, during epithelial-mesenchymal transition. Journal of Biological Chemistry, 288(3), 1469–1479. doi:10.1074/jbc.M112.438341.
Kalluri, R., & Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. Journal of Clinical Investigation, 119(6), 1420–1428. doi:10.1172/jci39104.
Lamouille, S., Xu, J., & Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nature Reviews Molecular Cell Biology, 15(3), 178–196. doi:10.1038/nrm3758.
Zhang, S., Wang, X., Osunkoya, A. O., Iqbal, S., Wang, Y., Chen, Z., et al. (2011). EPLIN downregulation promotes epithelial-mesenchymal transition in prostate cancer cells and correlates with clinical lymph node metastasis. Oncogene, 30(50), 4941–4952. doi:10.1038/onc.2011.199.
Acknowledgments
The authors are grateful to Cancer Research Wales and the Welsh Life Science Network - Ser Cymru for supporting this work.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Collins, R.J., Jiang, W.G., Hargest, R. et al. EPLIN: a fundamental actin regulator in cancer metastasis?. Cancer Metastasis Rev 34, 753–764 (2015). https://doi.org/10.1007/s10555-015-9595-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-015-9595-8