
Abstract
African Americans have a disproportionate burden of aggressive young-onset breast cancer. Genomic testing for inherited predisposition to breast cancer is increasingly common in clinical practice, but comprehensive mutation profiles remain unknown for most minority populations. We evaluated 289 patients who self-identified as African American with primary invasive breast cancer and with personal or family cancer history or tumor characteristics associated with high genetic risk for all classes of germline mutations in known breast cancer susceptibility genes using a validated targeted capture and multiplex sequencing approach. Sixty-eight damaging germline mutations were identified in 65 (22 %, 95 % CI 18–28 %) of the 289 subjects. Proportions of patients with unequivocally damaging mutations in a breast cancer gene were 26 % (47/180; 95 % confident interval [CI] 20–33 %) of those with breast cancer diagnosis before age 45; 25 % (26/103; 95 % CI 17–35 %) of those with triple-negative breast cancer (TNBC); 29 % (45/156; 95 % CI 22–37 %) of those with a first or second degree relative with breast cancer before age 60 or with ovarian cancer; and 57 % (4/7; 95 % CI 18–90 %) of those with both breast and ovarian cancer. Of patients with mutations, 80 % (52/65) carried mutations in BRCA1 and BRCA2 genes and 20 % (13/65) carried mutations in PALB2, CHEK2, BARD1, ATM, PTEN, or TP53. The mutational allelic spectrum was highly heterogeneous, with 57 different mutations in 65 patients. Of patients meeting selection criteria other than family history (i.e., with young age at diagnosis or TNBC), 48 % (64/133) had very limited information about the history of cancer in previous generations of their families. Mutations in BRCA1 and BRCA2 or another breast cancer gene occur in one in four African American breast cancer patients with early onset disease, family history of breast or ovarian cancer, or TNBC. Each of these criteria defines patients who would benefit from genomic testing and novel therapies targeting DNA repair pathways.
Similar content being viewed by others


Introduction
African American breast cancer patients are more likely than breast cancer patients from other populations to be affected at a young age; to develop aggressive, triple-negative breast cancers (TNBC) that lack expression of estrogen, progesterone, and HER2 receptors; and to die from their disease [1–4]. Furthermore, patients from all populations with inherited mutations in BRCA1 (MIM 113705) and BRCA2 (MIM 600185) are more likely to be diagnosed at a young age, and especially among BRCA1 carriers, to develop TNBC [5, 6]. Thus, one potential contributor to the higher incidence of early-onset aggressive breast cancer among African American patients may be a previously undefined higher burden of inherited breast cancer in this population. High frequencies of mutations in BRCA1 and BRCA2 have already been observed in breast cancer patients of African ancestry from Nigeria [7] and from the Bahamas [8]. In addition to BRCA1 and BRCA2, other genes that harbor mutations that predispose to breast cancer include PALB2 [9, 10], CHEK2 [11], ATM [12–15], BARD1 [16, 17], RAD51C [18], RAD51D [19], TP53 [20], and PTEN [21]. The relationship between aggressive young-onset breast cancer and mutations in these other genes is not yet well characterized.
Advances in DNA sequencing technologies have yielded assays that efficiently and cost-effectively screen multiple genes at the same time. One such assay, BROCA, was designed and validated for the simultaneous detection of mutations in multiple genomic regions encompassing all known and emerging breast cancer susceptibility genes [22, 23]. BROCA detects, in a single assay, all classes of mutations: single nucleotide variants, small insertions and deletions (indels), and large genomic rearrangements.
To investigate the genomic basis of early-onset breast cancer in African American women, we examined the frequencies of inherited mutations in cancer genes in self-reported African American patients ascertained through a cancer genetics clinic at a single tertiary-care medical center.
Patients and methods
Patients
Participants were women who self-reported as African American, who were diagnosed with primary invasive breast cancer, and who were ascertained at The University of Chicago Medicine Breast Program and the Cancer Risk Clinic between 1993 and July 2013. The University of Chicago serves a population catchment area that is predominantly African American; approximately 35 % of ethnically identified samples in our Breast Program biospecimen bank are derived from African Americans. Implementation of dedicated consenting led to a remarkably high consent rate of nearly 95 % on our biospecimen banking protocol. If related affected persons appeared on the registry, only the first to be listed was included. Of 502 unrelated African American women with a diagnosis of invasive breast cancer, DNA was available from 370 patients. Among them, a total of 289 African American patients were included in the present study based on meeting at least one of the following criteria: breast cancer diagnosis before age 45; personal history of both breast and ovarian cancer; TNBC; or family history of breast cancer before age 60 or of ovarian cancer at any age in a first or second degree relative. The Institutional Review Boards (IRB) at The University of Chicago and the University of Washington approved the study. Patients had been consented for use of their samples for future genomic analysis. Permission for use for this study was obtained from the IRB. BROCA testing results were returned to participants according to their preference of knowing or omitting the information.
Mutation detection
From each subject, 3 ug of DNA were used to prepare paired-end libraries with 150-bp inserts, which were hybridized to a custom pool of oligonucleotides designed to capture 48 genomic regions, and sequenced as previously described [22, 23]. For this project, only unambiguously damaging mutations in breast cancer genes BRCA1, BRCA2, PALB2, CHEK2, ATM, BARD1, RAD51C, RAD51D, TP53, and PTEN were evaluated. Single base-pair substitutions and indels [23] and large copy number variants (CNVs) [24] were identified as previously described. Unambiguously damaging mutations were defined as truncations from any source, complete gene deletions, splice mutations shown experimentally to yield truncations, and missense mutations shown experimentally to disrupt protein function. Single base-pair substitutions and indels were validated by independent PCR amplification and Sanger sequencing. CNVs were validated by real-time PCR using TaqMan probes (Life Technologies, Carlsbad, CA, USA) targeting the deleted or duplicated region. Qualifying patients were included in the study regardless of previous genetic testing; for those with prior genetic testing, sequencing and analysis was repeated, blind to previous results.
Statistic analysis
Proportions of mutation carriers and exact 95 % confidence intervals (CI) were estimated using binominal distribution. Fisher’s exact tests were used to examine whether mutation proportions vary across age groups or among patients meeting different numbers of eligibility criteria.
Results
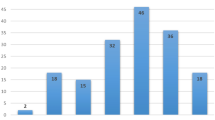
A total of 289 patients qualified for the study; their clinical features are shown in Table 1. Approximately, half of qualifying patients met more than one selection criterion, as illustrated in Fig. 1a. BROCA yielded >400-fold median coverage with 94 % of targeted bases covered >50-fold and 99.8 % of bases covered >10-fold. Sixty-eight damaging mutations were identified in 65 (22 %, 95 % CI 18–28 %) of the 289 subjects. All mutations previously identified and published [25, 26] were independently identified by BROCA. The proportions of patients with unequivocally damaging mutations were 26 % (47/180; 95 % CI 20–33 %) of those with breast cancer diagnosis before age 45, 25 % (26/103; 95 % CI 17–35 %) of those with TNBC, 29 % (45/156; 95 % CI 22–37 %) of those with a qualifying family history, and 57 % (4/7; 95 % CI 18–90 %) of the small group with both breast and ovarian cancer (Fig. 1b).

Frequencies of inherited mutations in breast cancer genes among African American breast cancer patients with young age at diagnosis, family history of breast or ovarian cancer, or triple negative breast cancer (TNBC). a Numbers of patients meeting each of the study criteria. All patients were diagnosed with primary invasive breast cancer and self-identified as African American. FH+ indicates a first or second degree relative with breast cancer diagnosed at age <60 or ovarian cancer at any age; Br/Ov indicates breast and ovarian cancer in the same subject; Br dx <45 indicates that the participant’s breast cancer was diagnosed at age <45; TNBC indicates that the breast cancer was triple negative; i.e., lacking estrogen and progesterone receptors and Her2. The study included 289 patients in all. b Proportions of patients with an unequivocally damaging mutation in a breast cancer gene, for each group from (a)
The mutations were distributed among eight genes, with the most in BRCA1 (29) and BRCA2 (23) and the remaining in PALB2 (3), ATM (5), CHEK2 (4), BARD1 (1), TP53 (1), and PTEN (2) (Fig. 2). No truncations were present in either RAD51C or RAD51D, even among the 35 patients with personal or family history of ovarian cancer. Four mutations were large genomic deletions, three in BRCA1 and one in ATM. All damaging mutations are indicated in Table 2. In addition to the 68 damaging mutations, we classified two variants as unknown significance, or “VUS”: BRCA1_5160A >G (U14680) in exon 17, and BRCA2_8614C >T (U43746) in exon 19, both predicted to alter splicing by disrupting exonic splice enhancer sites, but thus far without functional tests of patient RNA. These two variants were not included in subsequent analyses.

Eight genes with mutations in African American breast cancer patients. A total of 68 mutations were identified, 76 % in BRCA1 or BRCA2 and 24 % in other breast cancer genes
Patients meeting more than one qualifying criteria were more likely to carry a mutation (P = 0.001). Proportions of patients with mutations were 15 % (22/144) of those meeting only one criterion, 25 % (29/116) of those meeting two criteria, and 48 % (14/29) of those meeting three criteria. No patients met all four criteria. The distribution of mutations gene-by-gene differed in that mutations in BRCA1 were more frequent among patients diagnosed at age <45 than among patients diagnosed at age ≥45 and selected on other criteria (24/180 versus 5/109; P = 0.016). There were no other differences in the distribution of mutations gene-by-gene among patients selected by various criteria. The prevalence of damaging mutations among familial TNBC cases was 34.0 % (16/47) versus 17.9 % (10/56) in non-familial TNBC, but this was not statistically significant (P = 0.071).
Three patients carried two mutations each. Patient 101 was homozygous for CHEK2 p.G210R and was diagnosed with TNBC at age 33. Patient 276 carried both BRCA2 p.R3128X and ATM p.R248X. She was diagnosed with receptor-positive breast cancer at age 31. Patient 312 carried both CHEK2 p.R160G and ATM p.R457X. She was diagnosed with stage 3, receptor-positive breast cancer at age 34.
Three patients carried mutations in genes responsible for breast cancer in the context of syndromic disease. Patient 446 carried TP53 p.P278A. She was diagnosed at ages 32 and 36 with bilateral breast cancer. Her family history is unknown. Patient 333 carried PTEN p.R335X. She has hyperkeratosis and was diagnosed at age 35 with bilateral breast cancer, including foci of TNBC and DCIS and LCIS, and at age 46 with renal cell cancer. Her father carried the same mutation and was previously diagnosed with Cowden syndrome (MIM 158350). He developed renal cell cancer at age 53. The family includes multiple other relatives with cancer. Patient 248 carried PTEN c.1027(ivs8)(-1)G>T, which alters splicing of the last exon of PTEN, leading to reduced stability of the PTEN message. She was diagnosed with breast cancer at age 43; her family cancer history is unknown.
Discussion
These results are based on the largest clinic-based cohort of prospectively ascertained African American breast cancer patients to undergo genomic testing of cancer genes known to be associated with inherited breast cancer. Using a more cost-effective and innovative high-throughput sequencing technology, we found that nearly one in four of patients meeting selection criteria carried an inherited damaging mutation in at least one gene. There were 57 different pathogenic mutations in eight different genes among 65 patients, supporting the clinical utility of simultaneous multi-gene testing, rather than relying on a limited mutation panel or a gene-by-gene approach. The majority of mutations were found in BRCA1 and BRCA2 genes. These results are consistent with data from clinic-based cohorts in other populations.
There are strengths and weaknesses to any single institution study. There is paucity of data on well-phenotyped patients of African ancestry. This study represents the first report of a large cohort of self-reported African American patients with integrated mutation results from next-generation sequencing, clinical characteristics of the patients, and tumor phenotype. Our results confirm that inherited mutations in the BRCA1 and BRCA2 tumor suppressor genes are still the strongest predictors of breast and/or ovarian cancer risk in women of African ancestry. It is well recognized that prevalence of mutations in both genes among breast cancer patients vary by ethnicity, study inclusion criteria, and mutation detection techniques [27]. In this study, we found 10.0 % (29 out of 289) of cases carried pathogenic BRCA1 mutations and 8.0 % (23 out of 289) of cases are BRCA2-positive. The prevalence of recurrent BRCA1 and BRCA2 mutations among selected breast cancer patients are higher in founder populations such as Ashkenazi Jewish populations where a relatively cost-effective strategy of testing for three common mutations in BRCA1 and BRCA2 genes currently exists [25, 28, 29]. In a study examining BRCA1, BRCA2, CHEK2, and TP53 in US high-risk families (95 % were of European ancestry), 9.6 and 6.5 % of probands were found to be BRCA1 and BRCA2 mutation carriers, and, mutation prevalence increased to 13.2 % (BRCA1) and 7.4 % (BRCA2, CHEK2 and TP53) among patients <49 years old, respectively [30]. We have previously reported high mutation rates among clinic-based cohorts of women of African ancestry with early-onset breast cancer (≤45 years) [25]. In a population-based study from North California Breast Cancer Family Registry, BRCA1 mutation prevalence were 1.3, 2.2 and 3.5 % in African American, non-Hispanic and Hispanic patients overall but 16.7 % in African American cases diagnosed under the age of 35 years old [28]. Among 46,276 subjects (78.3 % Western European ancestry and 3.8 % African ancestry) tested by Myriad Genetics, Inc., BRCA1 and BRCA2 mutation prevalence was 10.2 and 5.7 % in the African ancestry group, respectively versus 6.9 and 5.2 % in populations of Western European ancestry [31]. In a genetic counseling TNBC cohort, BRCA1 and BRCA2 mutation prevalence has been reported to differ by populations [32]; 50 % in Ashkenazi Jewish women, 33.3 % in Caucasian women, and 20.4 % in African American women which is consistent with what we found in this study. A limitation of our study is that there is significant genetic diversity in the African Diaspora and results from our single institution study are not generalizable to the general population. Larger studies using next-generation sequencing in diverse populations are needed to derive true estimates of the burden of inherited breast cancer in underserved and understudied populations.
Next-generation sequencing assays promise to accelerate progress in screening for somatic as well as inherited genetic mutations among cancer patients. Several laboratories now offer genomic testing with reasonable turn around times, but the clinical utility of these cancer gene panels remain in question. BRCAplus, a high-risk breast cancer diagnostic assay, includes six breast cancer genes: BRCA1, BRCA2, TP53, PTEN, STK11, and CDH1 [33] and has been evaluated in more than 3,000 clinical samples referred to Ambry Genetics for mutation detection. The result showed that BRCA1 and BRCA2 mutations were found in 5.7 % of the patients and contributed 85 % of all the mutations identified. Another 27 gene targeted sequencing panel has also been evaluated in 708 hereditary breast and/or ovarian cancer patients [34]. In total, 109 germline mutations were detected, among them, 37 in BRCA1, 32 in BRCA2, 4 in TP53, and 36 in other genes. Similarly, BRCA1 and BRCA2 mutations accounted for around 60 % of the positive test results. Using BROCA in this study, 80 % of the cases had mutations in BRCA1 and BRCA2 while mutations in PALB2, CHEK2, BARD1, ATM, PTEN, and TP53 were found in the remaining 20 %. Given the diversity in ethnic populations, mutation detection methods, breast cancer genes included, degree of disease risk, sample size, and so on, the estimate of the prevalence of mutations in the rapidly expanding number of cancer susceptibility genes will also vary. Nonetheless, these massively parallel sequencing panels indeed prove their useful implementation in clinics and confirm that genes involved in DNA repair pathways, including BRCA1 and BRCA2 are the major genes contributing to inherited breast and ovarian cancer in African American populations [10].
The limited information about family history of cancer in several of these patients reflects a general problem. Of patients meeting selection criteria other than family history (i.e., selected for young age at diagnosis or TNBC), 48 % (64/133) had very limited information about cancers in previous generations of their families. This problem likely reflects historical disparities in access to medical care and strongly suggests that criteria other than family history should be widely recognized by clinicians and sufficient for referral for genomic testing. As has been demonstrated in Ashkenazi Jewish women and women of European ancestry [32], our results suggest that African American women with young age at onset, family history of breast or ovarian cancer, or TNBC could benefit very substantially from genomic testing. Identifying inherited mutations in breast cancer patients is important for at least three reasons. Patients with inherited mutations in genes involved in DNA repair pathways may be treated with synthetic lethal therapeutic approaches based on poly-ADP-ribose polymerase inhibitors (PARPi) [35–37]. In addition, women with inherited mutations in BRCA1, BRCA2, and PALB2 should be advised about risk-reducing salpingo-oophorectomy [38]. Furthermore, each breast cancer patient with inherited disease represents a family including as-yet-unaffected sisters and daughters and nieces for whom genetic knowledge can empower cancer prevention. Increased focus on cancer control and prevention to improve population health has the potential to reduce cost of care for advanced disease in underserved populations.
Despite its limitation as a single institution study, this study speaks to a fundamental problem of disparities in breast cancer outcomes among African American women. African American breast cancer patients are more likely than breast cancer patients of other ancestries to be affected at a young age; to develop aggressive breast cancers; and to die from their disease [1, 3, 4]. It is also known that patients with inherited mutations in BRCA1 are more likely to be diagnosed at a young age [5, 6]. The distribution of genetic and non-genetic risk factors for breast cancer is not uniform across populations and there is significant heterogeneity in breast cancer. It is possible that one potential contributor to the higher incidence of aggressive early-onset breast cancer among African American patients is a higher burden of previously understudied inherited mutations in breast cancer susceptibility genes. In order to test this hypothesis rigorously, taking into consideration the significant genetic diversity in the African Diaspora, it would be necessary to evaluate large, population-based cohorts of breast cancer patients of African and other ancestries, including both comprehensive genomic analysis and thorough characterization of patient history, tumor characteristics, treatment, and survival. Our study addressed the converse question: the frequency of mutations given family history or young-onset aggressive breast cancer. We suggest that the mutation frequency of approximately 25 % among African American patients with these features underscores the need for larger studies among women from diverse populations.
Heretofore, high cost and insurance coverage has been a major barrier to incorporating genomic testing into clinical cancer care. Targeted capture and multiplexed sequencing offers the capacity to sequence multiple relevant genes at once, in a single test, rather than having to order sequential genetic tests at a cost of thousands of dollars per gene. The cost of genetic testing for breast cancer predisposition should drop quickly in a competitive marketplace, allowing more equitable access. Diffusion of genetic technologies in diverse populations has the potential to accelerate precision medicine, improve cancer prevention, lower costs of cancer care, and reduce disparities in health outcomes.
References
Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC (2006) Race, breast cancer subtypes, and survival in the Carolina breast cancer Study. JAMA 295(21):2492–2502
Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, Perou CM, Nielsen TO (2008) Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res 14(5):1368–1376
DeSantis C, Ma J, Bryan L, Jemal A (2014) Breast cancer statistics, 2013. CA Cancer J Clin 64(1):52–62
Menashe I, Anderson WF, Jatoi I, Rosenberg PS (2009) Underlying causes of the black-white racial disparity in breast cancer mortality: a population-based analysis. J Natl Cancer Inst 101(14):993–1000
Atchley DP, Albarracin CT, Lopez A, Valero V, Amos CI, Gonzalez-Angulo AM, Hortobagyi GN, Arun BK (2008) Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J Clin Oncol 26(26):4282–4288
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363(20):1938–1948
Fackenthal JD, Zhang J, Zhang B, Zheng Y, Hagos F, Burrill DR, Niu Q, Huo D, Sveen WE, Ogundiran T, Adebamowo C, Odetunde A, Falusi AG, Olopade OI (2012) High prevalence of BRCA1 and BRCA2 mutations in unselected Nigerian breast cancer patients. Int J Cancer 131(5):1114–1123
Akbari MR, Donenberg T, Lunn J, Curling D, Turnquest T, Krill-Jackson E, Zhang S, Narod SA, Hurley J (2014) The spectrum of BRCA1 and BRCA2 mutations in breast cancer patients in the Bahamas. Clin Genet 85(1):64–67. doi:10.1111/cge.12132
Tischkowitz M, Capanu M, Sabbaghian N, Li L, Liang X, Vallee MP, Tavtigian SV, Concannon P, Foulkes WD, Bernstein L, Bernstein JL, Begg CB (2012) Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat 33(4):674–680
Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, Lee A, Subramanian D, De Leeneer K, Fostira F, Tomiak E, Neuhausen SL, Teo ZL, Khan S, Aittomaki K, Moilanen JS, Turnbull C, Seal S, Mannermaa A, Kallioniemi A, Lindeman GJ, Buys SS, Andrulis IL, Radice P, Tondini C, Manoukian S, Toland AE, Miron P, Weitzel JN, Domchek SM, Poppe B, Claes KB, Yannoukakos D, Concannon P, Bernstein JL, James PA, Easton DF, Goldgar DE, Hopper JL, Rahman N, Peterlongo P, Nevanlinna H, King MC, Couch FJ, Southey MC, Winqvist R, Foulkes WD, Tischkowitz M (2014) Breast-cancer risk in families with mutations in PALB2. N Engl J Med 371(6):497–506
Weischer M, Nordestgaard BG, Pharoah P, Bolla MK, Nevanlinna H, Van’t Veer LJ, Garcia-Closas M, Hopper JL, Hall P, Andrulis IL, Devilee P, Fasching PA, Anton-Culver H, Lambrechts D, Hooning M, Cox A, Giles GG, Burwinkel B, Lindblom A, Couch FJ, Mannermaa A, Alnaes GG, John EM, Dork T, Flyger H, Dunning AM, Wang Q, Muranen TA, van Hien R, Figueroa J, Southey MC, Czene K, Knight JA, Tollenaar RA, Beckmann MW, Ziogas A, Christiaens MR, Collee JM, Reed MW, Severi G, Marme F, Margolin S, Olson JE, Kosma VM, Kristensen VN, Miron A, Bogdanova N, Shah M, Blomqvist C, Broeks A, Sherman M, Phillips KA, Li J, Liu J, Glendon G, Seynaeve C, Ekici AB, Leunen K, Kriege M, Cross SS, Baglietto L, Sohn C, Wang X, Kataja V, Borresen-Dale AL, Meyer A, Easton DF, Schmidt MK, Bojesen SE (2012) CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer-specific death, and increased risk of a second breast cancer. J Clin Oncol 30(35):4308–4316
Goldgar DE, Healey S, Dowty JG, Da Silva L, Chen X, Spurdle AB, Terry MB, Daly MJ, Buys SM, Southey MC, Andrulis I, John EM, Khanna KK, Hopper JL, Oefner PJ, Lakhani S, Chenevix-Trench G (2011) Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res 13(4):R73
Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N (2006) ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 38(8):873–875
Tavtigian SV, Oefner PJ, Babikyan D, Hartmann A, Healey S, Le Calvez-Kelm F, Lesueur F, Byrnes GB, Chuang SC, Forey N, Feuchtinger C, Gioia L, Hall J, Hashibe M, Herte B, McKay-Chopin S, Thomas A, Vallee MP, Voegele C, Webb PM, Whiteman DC, Sangrajrang S, Hopper JL, Southey MC, Andrulis IL, John EM, Chenevix-Trench G (2009) Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Am J Hum Genet 85(4):427–446
Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF (2005) Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst 97(11):813–822
Baer R (2013) Luring BRCA1 to the scene of the crime. Cancer Cell 23(5):565–567
Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D 3rd, Fukuda M, Ohta T, Klevit R (2003) Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci USA 100(10):5646–5651
Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L, Schaal H, Ramser J, Honisch E, Kubisch C, Wichmann HE, Kast K, Deissler H, Engel C, Muller-Myhsok B, Neveling K, Kiechle M, Mathew CG, Schindler D, Schmutzler RK, Hanenberg H (2010) Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 42(5):410–414
Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, Bowden G, Kalmyrzaev B, Warren-Perry M, Snape K, Adlard JW, Barwell J, Berg J, Brady AF, Brewer C, Brice G, Chapman C, Cook J, Davidson R, Donaldson A, Douglas F, Greenhalgh L, Henderson A, Izatt L, Kumar A, Lalloo F, Miedzybrodzka Z, Morrison PJ, Paterson J, Porteous M, Rogers MT, Shanley S, Walker L, Eccles D, Evans DG, Renwick A, Seal S, Lord CJ, Ashworth A, Reis-Filho JS, Antoniou AC, Rahman N (2011) Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet 43(9):879–882
Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, Han JH, Lowstuter K, Longmate J, Sommer SS, Weitzel JN (2009) Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 27(8):1250–1256
Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C (2012) Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 18(2):400–407
Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A, Norquist B, Pennington KP, Garcia RL, King MC, Swisher EM (2011) Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA 108(44):18032–18037
Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, Nord AS, Mandell JB, Swisher EM, King MC (2010) Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA 107(28):12629–12633
Nord AS, Lee M, King MC, Walsh T (2011) Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genom 12:184
Nanda R, Schumm LP, Cummings S, Fackenthal JD, Sveen L, Ademuyiwa F, Cobleigh M, Esserman L, Lindor NM, Neuhausen SL, Olopade OI (2005) Genetic testing in an ethnically diverse cohort of high-risk women: a comparative analysis of BRCA1 and BRCA2 mutations in American families of European and African ancestry. JAMA 294(15):1925–1933
Zheng Y, Zhang J, Niu Q, Huo D, Olopade OI (2012) Novel germline PALB2 truncating mutations in African American breast cancer patients. Cancer 118(5):1362–1370
Fackenthal JD, Olopade OI (2007) Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer 7(12):937–948
John EM, Miron A, Gong G, Phipps AI, Felberg A, Li FP, West DW, Whittemore AS (2007) Prevalence of pathogenic BRCA1 mutation carriers in 5 US racial/ethnic groups. JAMA 298(24):2869–2876
Malone KE, Daling JR, Doody DR, Hsu L, Bernstein L, Coates RJ, Marchbanks PA, Simon MS, McDonald JA, Norman SA, Strom BL, Burkman RT, Ursin G, Deapen D, Weiss LK, Folger S, Madeoy JJ, Friedrichsen DM, Suter NM, Humphrey MC, Spirtas R, Ostrander EA (2006) Prevalence and predictors of BRCA1 and BRCA2 mutations in a population-based study of breast cancer in white and black American women ages 35–64 years. Cancer Res 66(16):8297–8308
Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, Higgins J, Roach KC, Mandell J, Lee MK, Ciernikova S, Foretova L, Soucek P, King MC (2006) Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 295(12):1379–1388
Hall MJ, Reid JE, Burbidge LA, Pruss D, Deffenbaugh AM, Frye C, Wenstrup RJ, Ward BE, Scholl TA, Noll WW (2009) BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer 115(10):2222–2233
Greenup R, Buchanan A, Lorizio W, Rhoads K, Chan S, Leedom T, King R, McLennan J, Crawford B, Marcom PK, Hwang ES (2013) Prevalence of BRCA mutations among women with triple-negative breast cancer (TNBC) in a genetic counseling cohort. Ann Surg Oncol 20(10):3254–3258
Chong HK, Wang T, Lu HM, Seidler S, Lu H, Keiles S, Chao EC, Stuenkel AJ, Li X, Elliott AM (2014) The validation and clinical implementation of BRCAplus: a comprehensive high-risk breast cancer diagnostic assay. PLOS One 9(5):e97408
Castera L, Krieger S, Rousselin A, Legros A, Baumann JJ, Bruet O, Brault B, Fouillet R, Goardon N, Letac O, Baert-Desurmont S, Tinat J, Bera O, Dugast C, Berthet P, Polycarpe F, Layet V, Hardouin A, Frebourg T, Vaur D (2014) Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet. doi:10.1038/ejhg.2014.16
Ashworth A (2008) A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol 26(22):3785–3790
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361(2):123–134
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376(9737):235–244
Finch AP, Lubinski J, Moller P, Singer CF, Karlan B, Senter L, Rosen B, Maehle L, Ghadirian P, Cybulski C, Huzarski T, Eisen A, Foulkes WD, Kim-Sing C, Ainsworth P, Tung N, Lynch HT, Neuhausen S, Metcalfe KA, Thompson I, Murphy J, Sun P, Narod SA (2014) Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J Clin Oncol 32(15):1547–1553
Acknowledgments
We thank the patients for their participation in this study. This work was supported by the National Institutes of Health grants R01CA157744, R01CA89085, P50CA125183, and K12CA139160; and by the Breast Cancer Research Foundation, the Ralph and Marion Falk Medical Research Trust, and Susan G. Komen for the Cure. M.C.K and O.I.O. are American Cancer Society Professors.
Conflict of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Jane E. Churpek and Tom Walsh have contributed equally to the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Churpek, J.E., Walsh, T., Zheng, Y. et al. Inherited predisposition to breast cancer among African American women. Breast Cancer Res Treat 149, 31–39 (2015). https://doi.org/10.1007/s10549-014-3195-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-014-3195-0