
Abstract
The aromatase inhibitors exemestane and anastrozole are approved in Japan for first-line treatment of postmenopausal patients with advanced, hormone-receptor-positive breast cancer. This phase 3, randomized, double-blind study directly compared time to progression (TTP) for exemestane and anastrozole therapy in this patient population. Eligible patients were randomized to receive exemestane 25 mg or anastrozole 1 mg, each once daily. The primary endpoint was TTP based on assessment by an expert radiologic images review committee (ERIRC). Secondary endpoints included investigator-assessed TTP, time to treatment failure, overall survival, objective response rate, clinical benefit rate, and safety. A total 298 patients were randomized to receive exemestane (n = 149; mean age 63.4 years) or anastrozole (n = 149; mean age 64.0 years). Median ERIRC-assessed TTP was 13.8 and 11.1 months (hazard ratio = 1.007; 95 % confidence interval [CI]: 0.771, 1.317) and median investigator-assessed TTP was 13.8 and 13.7 months (hazard ratio = 1.059; 95 % CI: 0.816, 1.374) in the exemestane and anastrozole arms, respectively. Median overall survival was 60.1 months in the anastrozole arm and was not reached in the exemestane arm at data cutoff. The objective response rate was 43.9 % (95 % CI: 35.3, 52.8) and 39.1 % (95 % CI: 30.6, 48.1) in the exemestane and anastrozole arms, respectively. Treatment-related adverse events grade ≥3 occurred in 9.4 and 6.0 % of patients, and treatment-related serious adverse events occurred in 4.0 and 3.4 % of patients in the exemestane and anastrozole arms, respectively. In this study, the efficacy and safety profiles of exemestane were similar to those of anastrozole in Japanese patients with advanced, hormone-receptor-positive breast cancer; however, TTP non-inferiority of exemestane versus anastrozole was not confirmed.
Similar content being viewed by others

Introduction
Breast cancer is the most common invasive cancer in women, accounting for roughly 232,000 new cases and 40,000 deaths annually, or approximately 15 % of cancer-related deaths in women in the United States [1]. In Japan, the incidence of breast cancer increased from roughly 11,000 to 31,000 cases between 1975 and 1995, and is expected to increase to 50,000 cases by 2015 [2].
Most breast cancers (60 %) express estrogen receptor (ER) or progesterone receptor (PgR) and are responsive to estrogens for growth and proliferation [3]. Therefore, hormone-receptor-positive breast cancer can be treated by either blocking the ER with agents such as the selective ER modulator (SERM) tamoxifen, or by reducing the production of estrogens with aromatase inhibitors (AIs) [4–6].
Exemestane, a steroidal, irreversible AI structurally related to androstenedione, and anastrozole, a triazolic non-steroidal AI, are both approved in Japan for treatment of breast cancer in postmenopausal women. Results of studies directly comparing the efficacy and safety of AIs to tamoxifen or megestrol acetate for treatment of hormone-receptor-positive breast cancer in postmenopausal women have been extensively reported and indicate an overall favorable efficacy and safety profile for AIs compared with other agents [7–18]; however, only a limited number of studies have directly compared the efficacy and safety of different AIs (studies MA.27 [19, 20] and ACOSOG Z1031 [21]), and these were conducted in patients with early breast cancer. The current study was a multicenter, randomized, double-blind, non-inferiority study conducted in Japan to evaluate time to progression (TTP) in postmenopausal patients with hormone-receptor-positive advanced and/or recurrent breast cancer randomized to treatment with either exemestane or anastrozole. This is the first large-scale direct comparison of the safety and efficacy of different AIs for first-line treatment of patients with advanced breast cancer.
Methods
Patients
All patients provided written informed consent before participating in the trial. Postmenopausal patients at least 20 years of age with metastatic, progressive, or locally recurrent, inoperable, hormone-receptor-positive breast cancer confirmed histologically or cytologically at the time of primary tumor diagnosis or detection of metastasis were eligible. Enrollment ER and PgR status was determined at each participating site. When available, tumor tissues were further analyzed for some biomarkers as described below. Postmenopausal status was defined as no menstruation for 12 months before enrollment and follicle-stimulating hormone levels in the postmenopausal range. Patients who had 12 or more months of menopause induced by chemotherapy or luteinizing hormone-releasing hormone (LHRH) agonist and who were at least 45 years of age were also eligible. Additional eligibility requirements included Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1 and measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 or measureable bone lesions per Japanese Classification of Breast Cancer, Edition 14 [22, 23]. Patients who developed advanced or recurrent breast cancer during postoperative adjuvant hormonal therapy with anti-estrogen agents (e.g., tamoxifen) were also eligible; however, patients who received previous hormonal therapy (tamoxifen, LHRH agonists, ovariectomy), chemotherapy, or AI therapy for advanced breast cancer were not eligible. Patients with rapidly progressing disease, large-volume visceral disease, and brain metastases were also ineligible.
This study was conducted in compliance with the Declaration of Helsinki and its amendments and relevant International Conference on Harmonisation of Good Clinical Practice guidelines, and in agreement with the institutional boards.
Study design and treatment
This was a multicenter, randomized, double-blind, phase 3 study conducted at 64 sites. Eligible patients received 2 tablets of the study drugs (exemestane 25 mg plus anastrozole placebo or anastrozole 1 mg plus exemestane placebo) and were instructed to take the tablets orally once daily with food. Treatment began within 14 days of receiving informed consent and continued until disease progression, intolerable adverse event (AE), or death.
Patients were randomized to balance for major prognostic factors including site of disease, postoperative adjuvant tamoxifen therapy, bisphosphonate use (pamidronate and/or zoledronic acid), and study site. The database was to be locked and the study unblinded after 228 events (disease progression or breast cancer death) had occurred as assessed by the expert radiologic images review committee (ERIRC).
Endpoints and assessments
The primary endpoint was TTP, defined as the time from randomization to confirmed progressive disease or breast cancer death based on the ERIRC assessment. Patients last documented to be either alive, still on treatment or within 28 days of treatment discontinuation, or progression-free were censored at the date of the last objective disease assessment verifying lack of disease progression.
Secondary endpoints (based on RECIST version 1.0 criteria) included investigator-assessed TTP, overall survival (OS), objective response rate, clinical benefit rate, and time to treatment failure. Overall survival was defined as the time from the date of randomization to the date of death from any cause; patients last known to be alive were censored at the date of last contact. Time to treatment failure was defined as the time from the date of randomization to the date of first documentation of disease progression, symptomatic deterioration, death from any cause, or discontinuation of treatment due to AE, refusal, or other reason. Patients last known not to have failed treatment were censored at the date of the last visit that verified lack of treatment failure.
For patients with measurable disease, investigator- and ERIRC-assessed antitumor responses were assessed using RECIST version 1.0. For patients with measureable bone lesions, the Japanese Classification of Breast Cancer was used for assessments. Patients were assessed every 8 weeks up to week 48, every 12 weeks thereafter, and at the end of treatment or study withdrawal. Clinical benefit rate was defined as the proportion of patients who achieved complete response, partial response, or stable disease lasting at least 24 weeks.
The incidence and severity of AEs was assessed using Common Terminology Criteria for Adverse Events (CTCAE) version 3.0 and the Japanese translated version of Japan Clinical Oncology Group (JCOG) criteria for AEs.
Exploratory assessments included the effect of tumor growth-related factors present at the time of diagnosis of advanced/recurrent breast cancer on the efficacy of exemestane or anastrozole treatment.
Immunohistochemical assays of biomarkers
Immunohistochemical staining of formalin-fixed/paraffin-embedded tumor tissue samples, when available, was performed at a central laboratory (Department of Pathology, Tohoku University School of Medicine) [24] using antibodies to ER (ER1D5; Immunotech, Marseille, France), PgR (MAB429; Chemicon International Inc., Temecula, California, USA), HER2 (AO485; DakoCytomation Co. Ltd., Kyoto, Japan), Ki67 (MIB-1; DakoCytomation), and epidermal growth factor receptor (EGFR) (Pharma Dx, DakoCytomation). HER2 status was determined using the Hercep Test, and samples with a score of 3+ were considered HER2-positive. Samples were considered EGFR-positive if they contained tumor cells with EGFR staining along the entire circumference of the cells. Lastly, Ki67 status was determined by calculating the percentage of Ki67-positive cells in a sample of at least 1,000 carcinoma cells, and was reported as Ki67 labeling index.
Hematologic and bone marker assessment
Patient bone alkaline phosphatase and type I collagen cross-linked N telopeptide levels were assessed by chemiluminescent enzyme immunoassay and enzyme immunoassay, respectively. Patient high-density lipoprotein (HDL) cholesterol and low-density lipoprotein (LDL) cholesterol levels were assessed using enzymatic methods. All assays were performed by Mitsubishi BCL (currently, Mitsubishi Chemical Medience Corporation, Tokyo, Japan).
Statistical assessments
This study was designed to confirm the TTP non-inferiority (80 % power) of exemestane versus anastrozole as initial hormone therapy in postmenopausal women with advanced breast cancer in. A hazard ratio (HR) of 0.9 for TTP in favor of exemestane was hypothesized based on previous clinical data and the target population of this study [7, 14, 25, 26]. Non-inferiority of exemestane versus anastrozole was prospectively defined as an upper bound of the 95 % confidence interval (CI) for the exemestane versus anastrozole HR no higher than 1.25 based on results of previous studies [7, 14, 25, 26]. To obtain 80 % power in this setting, 222 events (disease progression or breast cancer-related death) were required.
Efficacy was analyzed for all patients in the full analysis set (FAS) or in the per protocol set (PPS). The FAS included patients who were randomized, received at least 1 dose of study medication, and had at least 1 efficacy evaluation. The PPS included a subset of patients from the FAS who satisfied all inclusion criteria, did not meet any exclusion criteria, did not violate concomitant medication criteria, and had a total drug adherence rate of at least 80 %. The FAS was the primary efficacy analysis population.
The TTP HR and 95 % CI for exemestane versus anastrozole were calculated using a Cox proportional hazard model adjusting for key covariates (site of disease, postoperative adjuvant tamoxifen therapy, bisphosphonate use, and study site). Secondary TTP HRs with 95 % CI were determined using non-adjusted data (for FAS) or both adjusted and non-adjusted data (for PPS). Median TTP with 95 % CI was estimated for each treatment group by the Kaplan–Meier method.
For the FAS and PPS analysis populations, antitumor response point estimates and 95 % CI for each treatment group and the group difference were calculated and used to determine the rates of response and clinical benefit. Point estimates and 95 % CI were likewise calculated for intergroup differences in tumor responses. Median OS, median time to treatment failure, and 95 % CI were calculated by the Kaplan–Meier method. Investigator-assessed TTP was calculated in the same manner as the primary endpoint.
Safety data were summarized for all treated patients using descriptive statistics.
Results
Patient demographics and baseline characteristics
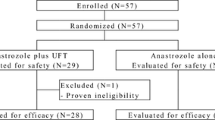
Patients were enrolled between April 20, 2005, and December 17, 2010. Demographics and baseline characteristics were well balanced between treatment groups (Table 1) [24]. A total 298 patients were randomly assigned to receive treatment with exemestane (n = 149; mean age 63.4, range 44–95 years) or anastrozole (n = 149; mean age 64.0, range 45–94 years). The FAS population included 147 (98.7 %) and 145 (97.3 %) patients from the exemestane and anastrozole groups, respectively, whereas the PPS population included 142 (95.3 %) and 138 (92.6 %) patients from the exemestane and anastrozole groups, respectively (Fig. 1). Results of immunohistochemical detection of ER, PgR, HER2, androgen receptor, EGFR, and Ki67 (from 115 patients in the exemestane arm and 110 patients in the anastrozole arm) determined at a central laboratory are presented in Table 1. The majority of patients with available histology data had tumors that stained positive for ER and/or PgR and negative for HER2.

Study flowchart. CT computed tomography, FAS full analysis set, MRI magnetic resonance imaging, PPS per protocol set
Efficacy
Median TTP in the FAS population based on ERIRC assessment was 13.8 months (95 % CI: 10.8, 16.5 months) and 11.1 months (95 % CI: 10.8, 16.6 months) in the exemestane and anastrozole groups, respectively (Table 2; Fig. 2a) [24]. The adjusted HR for TTP in the exemestane versus anastrozole groups was 1.007 (95 % CI: 0.771, 1.317). Non-inferiority was not confirmed because the upper limit in the 95 % CI (1.317) was larger than the prespecified non-inferiority margin (1.25).

Kaplan–Meier plots of a time to progression (ERIRC-assessed) and b overall survival. CI confidence interval, HR hazard ratio, NR not reached, OS overall survival, ERIRC expert radiologic images review committee, TTP time to progression. Reproduced with permission from Masuda et al. [24]
Additional secondary efficacy analyses demonstrated no significant differences between treatment groups. Although median OS was not reached in the exemestane group and was 60.1 months in the anastrozole group, the Kaplan–Meier plots indicated no difference in OS (Fig. 2b) [24]. Median time to treatment failure in the FAS population was 13.6 months (95 % CI: 9.2, 16.6 months) and 11.1 months (95 % CI: 9.4, 14.1 months) in the exemestane and anastrozole groups, respectively. Tumor response was evaluable for 132 and 128 patients in the exemestane and anastrozole groups, respectively. Complete response was reported in approximately 2 % of patients in both the exemestane (n = 2) and anastrozole (n = 3) groups, and partial response was reported in 42.4 % (n = 56) and 36.7 % (n = 47) of patients in the exemestane and anastrozole groups, respectively (Table 3) [24].
Safety
AEs from any cause were reported in 136 patients (91.3 %) in the exemestane group and 131 patients (87.9 %) in the anastrozole group, whereas treatment-related AEs occurred in 106 patients (71.1 %) in the exemestane group and 89 patients (59.7 %) in the anastrozole group (Table 4). Grade 3 or 4 AEs from any cause were reported in 28 patients (18.8 %) in the exemestane group and 27 patients (18.1 %) in the anastrozole group; grade 3 or 4 treatment-related AEs were reported in 13 patients (8.7 %) in the exemestane group and 9 patients (6.0 %) in the anastrozole group. The most common treatment-related AEs in either group were hot flush, arthralgia, and musculoskeletal stiffness (Table 5) [24].
Serious AEs were reported in 19 patients (12.8 %) in each treatment group; serious AEs occurring in 2 or more patients were cataract and dyspnea (n = 2 each) in the exemestane group, and vomiting (n = 3), vertigo, pneumonia, and decreased appetite (n = 2 each) in the anastrozole group.
Overall, 10 patients (6.7 %) in the exemestane group and 9 patients (6.0 %) in the anastrozole group discontinued study treatment because of AEs. Abnormal hepatic function (n = 2 in the exemestane group) was the only AE leading to discontinuation in more than 1 patient. As shown in Fig. 1, 5 patients (2 from the exemestane group and 3 from the anastrozole group) did not have an efficacy assessment, but these patients were included in the safety analyses. None of these patients had a treatment-related serious AE.
There were 3 deaths in the exemestane group (acute myocardial infarction, metastases to lung, and breast cancer; n = 1 each) and 1 death in the anastrozole group (general physical health deterioration) as the result of AEs. Of these, only the acute myocardial infarction in the exemestane group was considered related to treatment.
No significant differences in laboratory test abnormalities were observed between treatment groups. Common grade 3 or higher laboratory test abnormalities (>5 %) included gamma-glutamyl transferase (GGT) increase (9.4 %) and hyperglycemia (5.4 %) in the exemestane group and GGT increase (7.4 %), hypoalbuminemia (6.0 %), and hyperglycemia (5.4 %) in the anastrozole group (Table 6).
Bone markers increased slightly in both treatment groups throughout the observation period (Table 7). No substantial change in total cholesterol, HDL-cholesterol, or LDL-cholesterol was observed in either treatment group; however, triglyceride was slightly decreased in the exemestane group.
Discussion
In this study in postmenopausal women with advanced or recurrent breast cancer, TTP was similar with exemestane or anastrozole treatment (adjusted HR = 1.007; 95 % CI, 0.771, 1.317). However, the upper limit of the 95 % CI was above the prespecified non-inferiority margin of 1.25. No significant differences in investigator-assessed TTP, objective response rate, and clinical benefit rate between the exemestane and anastrozole treatment groups were observed in this study.
The results of this study are comparable to those reported in study NCIC CTG MA.27 comparing exemestane and anastrozole in the adjuvant setting for patients with early stage breast cancer, in which there was no statistically significant difference in event-free survival in the overall (HR = 1.02; P = 0.85), node-negative (HR = 1.04; P = 0.73), or node-positive (HR = 0.99; P = 0.90) populations [19, 20, 27]. Likewise, median TTP in the exemestane (13.8 months) or anastrozole (11.1 months) groups in the current study was comparable to median TTP for exemestane in the overall (9.9 months) [16] and Japanese (10.9 months) [28] populations, respectively, in the EORTC 10951 phase 3 study in postmenopausal women with metastatic breast cancer, as well as historical TTP data reported for anastrozole and letrozole [7, 10, 12, 14, 17]. Therefore, data from the current study are consistent with other reports demonstrating no differences in efficacy measures between AIs used as first-line treatment in patients with hormone-receptor-positive breast cancer.
The incidence of treatment-related AEs was higher in the exemestane group (71 %) versus the anastrozole group (60 %); however, most AEs were grade 1 or grade 2. The incidence of common AEs reported in this trial, including hot flush, arthralgia, and musculoskeletal stiffness, was consistent with previous comparisons of exemestane and anastrozole that found few differences between these agents in the adjuvant setting [19, 29].
Observed changes in plasma lipoproteins and bone markers were minimal between the two treatment groups in the current study. As reported in a recent systematic review, treatment with AIs did not correlate with any definitive change or unfavorable effect on plasma lipoproteins [30], and in a sub-study of Japanese women from the Tamoxifen Exemestane Adjuvant Multinational (TEAM) trial, exemestane or anastrozole treatment had no clinically significant effect on serum lipids in postmenopausal women with hormone-receptor-positive breast cancer [31]. Musculoskeletal symptoms and decreased bone mineral density are anticipated effects of hormonal therapies, such as AIs, that produce menopause-like effects [32]. In study MA.27, arthralgia, muscle pain, and fractures were reported in both the exemestane and anastrozole treatment groups; however, exemestane was associated with a significantly lower incidence of self-reported new-onset osteoporosis compared with anastrozole (P = 0.001) [20, 29]. Treatment algorithms for musculoskeletal AEs related to AI therapy have been developed, and discontinuation of AI therapy is rarely required [33–35].
In the present study, there was 1 death from acute myocardial infarction that was considered related to exemestane treatment, and no treatment-related deaths in the anastrozole group. Although results from two meta-analyses reported a higher risk of cardiovascular AEs associated with AIs compared with tamoxifen, these data have not been confirmed in a placebo-controlled trial, and available data do not support a substantial risk of ischemic cardiovascular events associated with AI treatment or differences in the risk of cardiovascular events for the different AIs [29, 30, 36–39]. Studies with longer follow-up time are needed to further define the cardiovascular safety profile of AI therapy.
Treatment guidelines have indicated no preference among approved AIs for patients with early breast cancer, and no efficacy or safety differences among AIs have been demonstrated in studies conducted in patients with advanced breast cancer [20, 40, 41]. Additional studies may help identify prognostic factors or predictive markers to help provide optimized treatment for the individual patient. Several additional clinical trials directly comparing exemestane to other AIs in patients with advanced cancer are in progress and may yield additional information to aid in treatment decisions [21, 42, 43].
Abbreviations
- AE:
-
Adverse event
- AI:
-
Aromatase inhibitor
- CI:
-
Confidence interval
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- ECOG:
-
Eastern Cooperative Oncology Group
- EGFR:
-
Epidermal growth factor receptor
- ER:
-
Estrogen receptor
- ERIRC:
-
Expert radiologic images review committee
- FAS:
-
Full analysis set
- GGT:
-
Gamma-glutamyl transferase
- HDL:
-
High-density lipoprotein
- HER2:
-
Human epidermal growth factor
- HR:
-
Hazard ratio
- JCOG:
-
Japan Clinical Oncology Group
- LDL:
-
Low-density lipoprotein
- LHRH:
-
Luteinizing hormone-releasing hormone
- OS:
-
Overall survival
- PgR:
-
Progesterone receptor
- PPS:
-
Per protocol set
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- SERM:
-
Selective ER modulator
- TTP:
-
Time to progression
References
American Cancer Society (2011) Cancer Facts & Figures 2011. American Cancer Society. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-029771.pdf. Accessed 4 October 2012
Ohno Y, Nakamura T, Murata K, Tsukuma H, Ajiki W, Oshima A (2004) Prediction of cancer incidence in Japan. In: Oshima A, Kuroishi T, Tajima K (eds) Cancer Statistics-2004. Shinohara Shuppan, Tokyo, pp 201–217
Anderson WF, Pfeiffer RM, Dores GM, Sherman ME (2006) Comparison of age distribution patterns for different histopathologic types of breast carcinoma. Cancer Epidemiol Biomarkers Prev 15(10):1899–1905. doi:10.1158/1055-9965.EPI-06-0191
Bhatnagar AS (2007) The discovery and mechanism of action of letrozole. Breast Cancer Res Treat 105(Suppl 1):7–17
Johnston SR (2005) Endocrinology and hormone therapy in breast cancer: selective oestrogen receptor modulators and downregulators for breast cancer—have they lost their way? Breast Cancer Res 7(3):119–130
Lonning PE (2004) Aromatase inhibitors in breast cancer. Endocr Relat Cancer 11(2):179–189
Bonneterre J, Thurlimann B, Robertson JF, Krzakowski M, Mauriac L, Koralewski P, Vergote I, Webster A, Steinberg M, von Euler M (2000) Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: results of the Tamoxifen or Arimidex Randomized Group Efficacy and Tolerability study. J Clin Oncol 18(22):3748–3757
Buzdar A, Douma J, Davidson N, Elledge R, Morgan M, Smith R, Porter L, Nabholtz J, Xiang X, Brady C (2001) Phase III, multicenter, double-blind, randomized study of letrozole, an aromatase inhibitor, for advanced breast cancer versus megestrol acetate. J Clin Oncol 19(14):3357–3366
Buzdar A, Jonat W, Howell A, Jones SE, Blomqvist C, Vogel CL, Eiermann W, Wolter JM, Azab M, Webster A, Plourde PV (1996) Anastrozole, a potent and selective aromatase inhibitor, versus megestrol acetate in postmenopausal women with advanced breast cancer: results of overview analysis of two phase III trials. Arimidex Study Group. J Clin Oncol 14(7):2000–2011
Dombernowsky P, Smith I, Falkson G, Leonard R, Panasci L, Bellmunt J, Bezwoda W, Gardin G, Gudgeon A, Morgan M, Fornasiero A, Hoffmann W, Michel J, Hatschek T, Tjabbes T, Chaudri HA, Hornberger U, Trunet PF (1998) Letrozole, a new oral aromatase inhibitor for advanced breast cancer: double-blind randomized trial showing a dose effect and improved efficacy and tolerability compared with megestrol acetate. J Clin Oncol 16(2):453–461
Kaufmann M, Bajetta E, Dirix LY, Fein LE, Jones SE, Zilembo N, Dugardyn JL, Nasurdi C, Mennel RG, Cervek J, Fowst C, Polli A, di Salle E, Arkhipov A, Piscitelli G, Miller LL, Massimini G (2000) Exemestane is superior to megestrol acetate after tamoxifen failure in postmenopausal women with advanced breast cancer: results of a phase III randomized double-blind trial. The Exemestane Study Group. J Clin Oncol 18(7):1399–1411
Mouridsen H, Gershanovich M, Sun Y, Perez-Carrion R, Boni C, Monnier A, Apffelstaedt J, Smith R, Sleeboom HP, Jaenicke F, Pluzanska A, Dank M, Becquart D, Bapsy PP, Salminen E, Snyder R, Chaudri-Ross H, Lang R, Wyld P, Bhatnagar A (2003) Phase III study of letrozole versus tamoxifen as first-line therapy of advanced breast cancer in postmenopausal women: analysis of survival and update of efficacy from the International Letrozole Breast Cancer Group. J Clin Oncol 21(11):2101–2109. doi:10.1200/JCO.2003.04.194
Nabholtz JM, Bonneterre J, Buzdar A, Robertson JF, Thurlimann B (2003) Anastrozole (Arimidex) versus tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: survival analysis and updated safety results. Eur J Cancer 39(12):1684–1689. doi:S0959804903003265
Nabholtz JM, Buzdar A, Pollak M, Harwin W, Burton G, Mangalik A, Steinberg M, Webster A, von Euler M (2000) Anastrozole is superior to tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: results of a North American multicenter randomized trial. Arimidex Study Group. J Clin Oncol 18(22):3758–3767
Paridaens R, Dirix L, Lohrisch C, Beex L, Nooij M, Cameron D, Biganzoli L, Cufer T, Duchateau L, Hamilton A, Lobelle JP, Piccart M (2003) Mature results of a randomized phase II multicenter study of exemestane versus tamoxifen as first-line hormone therapy for postmenopausal women with metastatic breast cancer. Ann Oncol 14(9):1391–1398
Paridaens RJ, Dirix LY, Beex LV, Nooij M, Cameron DA, Cufer T, Piccart MJ, Bogaerts J, Therasse P (2008) Phase III study comparing exemestane with tamoxifen as first-line hormonal treatment of metastatic breast cancer in postmenopausal women: the European Organisation for Research and Treatment of Cancer Breast Cancer Cooperative Group. J Clin Oncol 26(30):4883–4890. doi:10.1200/JCO.2007.14.4659
Thurlimann B, Hess D, Koberle D, Senn I, Ballabeni P, Pagani O, Perey L, Aebi S, Rochlitz C, Goldhirsch A (2004) Anastrozole (‘Arimidex’) versus tamoxifen as first-line therapy in postmenopausal women with advanced breast cancer: results of the double-blind cross-over SAKK trial 21/95—a sub-study of the TARGET (Tamoxifen or ‘Arimidex’ Randomized Group Efficacy and Tolerability) trial. Breast Cancer Res Treat 85(3):247–254. doi:10.1023/B:BREA.0000025420.78346.f9
van de Velde CJ, Rea D, Seynaeve C, Putter H, Hasenburg A, Vannetzel JM, Paridaens R, Markopoulos C, Hozumi Y, Hille ET, Kieback DG, Asmar L, Smeets J, Nortier JW, Hadji P, Bartlett JM, Jones SE (2011) Adjuvant tamoxifen and exemestane in early breast cancer (TEAM): a randomised phase 3 trial. Lancet 377(9762):321–331. doi:10.1016/S0140-6736(10)62312-4
Goss PE, Ingle JN, Chapman JAW, Ellis MJ, Sledge GW, Budd GT, Rabaglio M, Gelmon K, Shepherd L, Pritchard KI (2010) Final analysis of NCIC CTG MA.27: a randomized phase III trial of exemestane versus anastrozole in postmenopausal women with hormone receptor positive primary breast cancer. Cancer Res 70(24 Suppl 2):Abstract S1-1
Goss PE, Ingle JN, Pritchard KI, Ellis MJ, Sledge GW, Budd GT, Rabaglio M, Ansari RH, Johnson DB, Tozer R, D’Souza DP, Chalchal H, Spadafora S, Stearns V, Perez EA, Liedke PE, Lang I, Elliott C, Gelmon KA, Chapman JA, Shepherd LE (2013) Exemestane versus anastrozole in postmenopausal women with early breast cancer: NCIC CTG MA.27—a randomized controlled phase III trial. J Clin Oncol. doi:10.1200/JCO.2012.44.7805
Ellis MJ, Suman VJ, Hoog J, Lin L, Snider J, Prat A, Parker JS, Luo J, DeSchryver K, Allred DC, Esserman LJ, Unzeitig GW, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Watson MA, Leitch M, Hunt K, Olson JA (2011) Randomized phase II neoadjuvant comparison between letrozole, anastrozole, and exemestane for postmenopausal women with estrogen receptor-rich stage 2 to 3 breast cancer: clinical and biomarker outcomes and predictive value of the baseline PAM50-based intrinsic subtype-ACOSOG Z1031. J Clin Oncol 29(17):2342–2349. doi:10.1200/JCO.2010.31.6950
Japanese Breast Cancer Society (2000) General rules for clinical and pathological recording of breast cancer, 14th edn. Kanehara, Tokyo
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92(3):205–216
Masuda N, Iwata H, Ohno S, Rai S, Sato Y, Ohsumi S, Hashigaki S, Nishizawa Y, Saeki T, Noguchi S (2011) A randomized, double-blind, controlled study of exemestane versus anastrozole for first-line treatment of postmenopausal Japanese women with hormone receptor-positive advanced breast cancer. Poster presented at the 34th Annual San Antonio Breast Cancer Symposium, San Antonio, Texas; December 6–10, Abstract P1-16-01.
Paridaens R, Dirix L, Beex L, Nooij M, Cufer T, Lohrisch C, Biganzoli L, Van Hoorebeeck I, Duchateau L, Lobelle JP, Piccart M (2000) Exemestane (Aromasin) is active and well tolerated as first-line hormonal therapy (HT) of metastatic breast cancer (MBC) patients (Pts): results of a randomized phase II trial. Proc Am Soc Clin Oncol 19:83a (Abstract 316)
Paridaens R, Dirix L, Beex L, Nooij M, Cufer T, Lohrisch C, Biganzoli L, van Hoorebeeck I, Duchateau L, Lobelle JP, Piccart M (2000) Promising results with exemestane in the first-line treatment of metastatic breast dancer: a randomized phase II EORTC trial with a tamoxifen control. Clin Breast Cancer 1(Suppl 1):S19–S21
Pal S (2010) Anastrazole, exemestane prove equivalent as initial adjuvant therapy [summary of Goss presentation at 2010 SABCS]. http://www.cancernetwork.com/conference-reports/sabcs2010/content/article/10165/1754922. Accessed 4 September 2012
Sano M (2006) Phase 3 randomized trial of exemestane and tamoxifen (EORTC10951)—analysis of Japanese patients. Presented at the 14th Annual Scientific Meeting of the Japanese Breast Cancer Society, Kanazawa, Japan, July 2006, Abstract O-252.
Kittaneh M, Gluck S (2011) Exemestane in the adjuvant treatment of breast cancer in postmenopausal women. Breast Cancer (Auckl) 5:209–226. doi:10.4137/BCBCR.S6234
Younus M, Kissner M, Reich L, Wallis N (2011) Putting the cardiovascular safety of aromatase inhibitors in patients with early breast cancer into perspective: a systematic review of the literature. Drug Saf 34(12):1125–1149. doi:10.2165/11594170-000000000-00000
Hozumi Y, Suemasu K, Takei H, Aihara T, Takehara M, Saito T, Ohsumi S, Masuda N, Ohashi Y (2011) The effect of exemestane, anastrozole, and tamoxifen on lipid profiles in Japanese postmenopausal early breast cancer patients: final results of National Surgical Adjuvant Study BC 04, the TEAM Japan sub-study. Ann Oncol 22(8):1777–1782. doi:10.1093/annonc/mdq707
Shapiro CL (2005) Aromatase inhibitors and bone loss: risks in perspective. J Clin Oncol 23(22):4847–4849. doi:10.1200/JCO.2005.03.903
Hadji P, Body JJ, Aapro MS, Brufsky A, Coleman RE, Guise T, Lipton A, Tubiana-Hulin M (2008) Practical guidance for the management of aromatase inhibitor-associated bone loss. Ann Oncol 19(8):1407–1416. doi:10.1093/annonc/mdn164
Thorne C (2007) Management of arthralgias associated with aromatase inhibitor therapy. Curr Oncol 14(Suppl 1):S11–S19
Younus J, Kligman L (2010) Management of aromatase inhibitor-induced arthralgia. Curr Oncol 17(1):87–90
Cuppone F, Bria E, Verma S, Pritchard KI, Gandhi S, Carlini P, Milella M, Nistico C, Terzoli E, Cognetti F, Giannarelli D (2008) Do adjuvant aromatase inhibitors increase the cardiovascular risk in postmenopausal women with early breast cancer? Meta-analysis of randomized trials. Cancer 112(2):260–267. doi:10.1002/cncr.23171
Ewer MS, Gluck S (2009) A woman’s heart: the impact of adjuvant endocrine therapy on cardiovascular health. Cancer 115(9):1813–1826. doi:10.1002/cncr.24219
Amir E, Seruga B, Niraula S, Carlsson L, Ocana A (2011) Toxicity of adjuvant endocrine therapy in postmenopausal breast cancer patients: a systematic review and meta-analysis. J Natl Cancer Inst 103(17):1299–1309. doi:10.1093/jnci/djr242
Ligibel JA, James O’Malley A, Fisher M, Daniel GW, Winer EP, Keating NL (2012) Risk of myocardial infarction, stroke, and fracture in a cohort of community-based breast cancer patients. Breast Cancer Res Treat 131(2):589–597. doi:10.1007/s10549-011-1754-1
Goldhirsch A, Ingle JN, Gelber RD, Coates AS, Thurlimann B, Senn HJ (2009) Thresholds for therapies: highlights of the St Gallen International Expert Consensus on the primary therapy of early breast cancer. Ann Oncol 20(8):1319–1329. doi:10.1093/annonc/mdp322
National Comprehensive Cancer Network (2011) NCCN Clinical Practice Guidelines in Oncology: Breast Cancer. Version 2.2001. http://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed 13 September 2012
Gruppo Italiano Mammella (2010) Study of anastrozole, letrozole, or exemestane with or without tamoxifen in treating postmenopausal women with hormone-responsive breast cancer that has been completely removed by surgery. http://clinicaltrials.gov/ct2/show/NCT00541086. Accessed 13 September 2012
Southwest Oncology Group (2011) Tamoxifen citrate, letrozole, anastrozole, or exemestane with or without chemotherapy in treating patients with invasive RxPONDER breast cancer. http://clinicaltrials.gov/ct2/show/NCT01272037. Accessed 4 September 2012
US Department of Health and Human Services National Institutes of Health, National Cancer Institute Cancer Therapy Evaluation Program, Division of Cancer Treatment and Diagnosis (2006) Common terminology criteria for adverse events v3.0 (CTCAE). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed 13 September 2012
Acknowledgments
We would like to thank all participating patients and their families as well as the investigators, study coordinators, and operations staff. Financial support this study was provided by Pfizer Japan. Medical editorial/writing assistance was provided by Bret A. Wing, PhD, at Accuverus, a division of ProEd Communications, Inc., and was funded by Pfizer Inc. We thank Katsumasa Kuroi (Breast Oncology, Tokyo Metropolitan Cancer and Infectious Diseases Center Komagome Hospital) and Yasuo Ohashi (Biostatistics, School of Public Health, University of Tokyo), who were members of the independent data monitoring committee, and Izoh Kimishima (Surgery, Northern Fukushima Medical Center), Hirotaka Iwase (Breast and Endocrine Surgery, School of Medicine Kumamoto University), and Ryo Tamura (Department of Diagnostic Radiology, Tohoku University Graduate School of Medicine), who were members of the expert radiologic images review committee.
Conflict of interest
Authors Nishizawa and Hashigaki are employees of Pfizer. Author Noguchi has received grant/research support and honoraria from Pfizer and AstraZeneca, and is an advisor for AstraZeneca. Author Hiraoka has received research funding from Varian Medical Systems and Mitsubishi Heavy Industries. Author Sasano has received an educational grant from Pfizer. All other authors declare that they have no conflict of interest.
Ethical standards
This study complied with the laws of the country in which it was conducted (Japan).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Iwata, H., Masuda, N., Ohno, S. et al. A randomized, double-blind, controlled study of exemestane versus anastrozole for the first-line treatment of postmenopausal Japanese women with hormone-receptor-positive advanced breast cancer. Breast Cancer Res Treat 139, 441–451 (2013). https://doi.org/10.1007/s10549-013-2573-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-013-2573-3